
Abstract
Aim:
The macrophage-mediated inflammatory response may contribute to the development of cancer, diabetes, atherosclerosis and septic shock. This study was to characterize several new compounds to suppress macrophage-mediated inflammation.
Methods:
Peritoneal macrophages from C57BL/6 male mice and RAW264.7 cells were examined. Anti-inflammatory activity was evaluated in the cells exposed to lipopolysaccharide (LPS). The mechanisms of the anti-inflammatory activity were investigated via measuring transcription factor activation in response to specific signals and via assaying the activities of the target kinases.
Results:
Of 7 candidate compounds tested, 8-(tosylamino)quinoline (8-TQ, compound 7) exhibited the strongest activities in suppressing the production of NO, TNF-α, and PGE2 in LPS-activated RAW264.7 cells and peritoneal macrophages (the IC50 values=1−5 μmol/L). This compound (1.25−20 μmol/L) dose-dependently suppressed the expression of the pro-inflammatory genes for iNOS, COX-2, TNF-α, and the cytokines IL-1β and IL-6 at the level of transcription in LPS-activated RAW264.7 cells. 8-TQ (20 μmol/L) significantly suppressed the activation of NF-κB and its upstream signaling elements, including inhibitor of κB (IκBα), IκBα kinase (IKK) and Akt in LPS-activated RAW264.7 cells. In in vivo experiments, oral administration of 20 and 40 mg/kg 8-TQ for 3 d significantly alleviated the signs of LPS-induced hepatitis and HCl/EtOH-induced gastritis, respectively, in ICR mice.
Conclusion:
8-TQ (compound 7) exerts significant anti-inflammatory activity through the inhibition of the Akt/NF-κB pathway, thus may be developed as a novel anti-inflammatory drug.
Similar content being viewed by others
Introduction
Macrophages, the terminally differentiated progeny of monocytes, are characterized by cell-surface markers of the inflammatory response, including toll-like receptors (TLRs), the Fc receptor, and the complement receptor. Although macrophages function primarily as phagocytes and antigen-presenting cells, they may also activate other immuno-regulatory cells, including neutrophils and T- and B-lymphocytes1,2, through the production of pro-inflammatory cytokines, such as interleukins (ILs) and tumor necrosis factor (TNF)-α; chemokines; and inflammatory mediators such as nitric oxide (NO) and prostaglandin E2 (PGE2). The activation and chemotactic migration of inflammatory and immune cells into tissues play essential roles in the defense against viral, bacterial, and fungal infections. Uncoupled from normal controls, however, components of the immune response may contribute to a variety of acute and chronic disorders, including cancer, diabetes, septic shock, autoimmune diseases, and atherosclerosis3,4,5. In these disorders, tissue-associated macrophages predominate among the cells that directly injure tissues. These considerations led us to seek to develop a drug that might suppress macrophage-mediated inflammation. A variety of in vitro and in vivo models of inflammatory disease have been used in drug-screening studies. Macrophages in these systems may be activated by treatment with ligands such as lipopolysaccharide (LPS), peptidoglycan, and poly(I:C)6.
Recent approaches to anti-inflammatory drug development have focused on key signaling proteins as targets and have tested compounds for activity against them. Previously targeted proteins include the transcription factors nuclear factor (NF)-κB and activator protein (AP)-1 and their upstream activating enzymes, including inhibitor of κB (IκBα), IκBα kinase (IKK), Akt, phosphoinositide-dependent kinase-1 (PDK1), phosphoinositide 3-kinase (PI3K), the tyrosine kinases Syk and Src, and enzymes in the mitogen-activated protein kinase (MAPK) cascade [extracellular signal-regulated kinase (ERK), c-Jun N-terminal kinase (JNK), and p38]. These proteins play critical roles in regulating pro-inflammatory gene expression.
BAY11-7082 is a representative IKK inhibitor that actively suppresses various inflammatory cytokines7, the induction of heme oxygenase-18 and ICAM-1 expression9 and may potentiate neutrophil apoptosis10. This compound may prove beneficial in the treatment of inflammatory conditions such as arthritis11. Because we did not initially identify this compound, however, we face restrictions in developing it further. We believe we can overcome such restrictions by using derivatives of the original compound. For this study, we selected seven commercially available compounds (1 through 7) based on structural similarity to BAY 11-7082. We evaluated the anti-inflammatory activities of these seven analogs and investigated their molecular mechanisms.
Materials and methods
Materials
Test compounds 1 through 7 were purchased from Sigma-Aldrich Co (St Louis, MO, USA) at greater than 95% purity. Sodium carboxymethylcellulose (NaCMC), polyethylene glycol 400, (3-4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), GM-CSF, and LPS (E coli 0111:B4) were also obtained from Sigma. LY294002 (LY), BAY11-7082 (BAY), U0126, and wortmannin were from Calbiochem (La Jolla, CA, USA). Luciferase constructs containing binding promoters for NF-κB and AP-1 were used as reported previously12,13. Enzyme immunoassay (EIA) kits and enzyme-linked immunosorbent assay (ELISA) kits for PGE2 and TNF-α were purchased from Amersham (Little Chalfont, Buckinghamshire, UK). Fetal bovine serum and RPMI-1640 medium were obtained from GIBCO (Grand Island, NY, USA). RAW264.7 cells were purchased from ATCC (Rockville, MD, USA). All other chemicals were of Sigma reagent grade. Phospho-specific or total antibodies to transcription factors (p65, p50, c-Jun, STAT-1, and c-Fos), ERK (extracellular signal-related kinase), p38, JNK (c-Jun N-terminal kinase), IκBα, IKKβ, Akt, p85/PI3K, γ-tubulin, β-actin, and non-receptor tyrosine kinases (Src and Syk) were obtained from Cell Signaling Technology Inc (Beverly, MA, USA).
Animals
C57BL/6 male mice (6–8 weeks old, 17–21 g) were obtained from Dae Han Bio Link Co Ltd, Chungbuk, Korea, and maintained in plastic cages under conventional conditions. Water and pellet diets (Samyang Corp, Daejeon, Korea) were available ad libitum. Studies were performed in accordance with the guidelines established by the Kangwon University Institutional Animal Care and Use Committee.
Preparation of peritoneal and bone marrow-derived macrophages
Peritoneal exudates were obtained from C57BL/6 male mice (7–8 weeks old, 17–21 g) by lavage 4 d after the intraperitoneal injection of 1 mL of sterile 4% thioglycolate broth (Difco Laboratories, Detroit, MI, USA) as reported previously14,15. After washing with RPMI-1640 medium containing 2% FBS, peritoneal macrophages (1×106 cells/mL) were plated in 100-mm tissue culture dishes for 4 h at 37 °C in a 5% CO2 humidified atmosphere. To prepare bone marrow-derived macrophages, femurs and tibias were isolated from mice, and the muscle was removed. Bones were cut with scissors at both ends and flushed with 5 mL of RPMI-1640 with a 25-gauge needle. Cells were seeded at a density of 2×105 nucleated bone marrow cells/cm2 in RPMI-1640 containing 10% FBS, 100 U/mL penicillin, 0.1 mg/mL streptomycin, 2 mmol/L L-glutamine, and 50 ng/mL GM-CSF in 6-well CellBIND plates (Corning Life Sciences, Lowell, MA, USA) containing 3 mL per well. After a 24-h incubation, the cells were rinsed three times with 3 mL of RPMI-1640 to remove non-adherent cells and cultured further with 3 mL of RPMI-1640 containing 10% FBS, 100 U/mL penicillin, 0.1 mg/mL streptomycin, 2 mmol/L L-glutamine, and 50 ng/mL GM-CSF, hereafter referred to as “complete medium.” The cell culture medium was replaced every 3 d with fresh complete medium. After 3 weeks in culture, experiments were performed in serum-free RPMI-1640 containing 50 ng/mL GM-CSF and additions as indicated.
Cell culture
Peritoneal macrophages and cell lines (RAW264.7 and HEK293 cells) were cultured with RPMI-1640 medium supplemented with 10% heat-inactivated fetal bovine serum (Gibco, Grand Island, NY, USA), glutamine, and antibiotics (penicillin and streptomycin) at 37 °C under 5% CO2. For each experiment, cells were detached with a cell scraper. At our experimental cell density (2×106 cells/mL), the proportion of dead cells was less than 1% according to trypan blue dye exclusion tests.
NO, PGE2, and TNF-α production
After the preincubation of RAW264.7 cells or peritoneal macrophages (1×106 cells/mL) for 18 h, the cells were pre-treated with test compounds 1 through 7 for 30 min and were then incubated with LPS (1 μg/mL) for 24 h. The inhibitory effects of the test compounds on NO, PGE2, and TNF-α production were determined by analyzing the NO, PGE2, and TNF-α levels with the Griess reagent and enzyme-linked immunosorbent assay (ELISA) kits, as described previously16,17,18.
Cell viability test
RAW264.7 cells (1×106 cells/mL) were preincubated for 18 h and were then incubated for 24 h following the addition of test compounds 1 through 7 to the cells. The cytotoxic effects were evaluated by the MTT assay19. At 3 h prior to culture termination, 10 μL of an MTT solution (10 mg/mL in phosphate buffered-saline, pH 7.4) was added, and the cells were returned to culture until the end of the experiment. Incubation was halted by the addition of 15% sodium dodecyl sulfate to each well to solubilize the formazan20. The absorbance at 570 nm (OD570–630 nm) was measured using a SpectraMax 250 microplate reader.
mRNA analysis by semiquantitative reverse-transcription polymerase chain reaction (RT-PCR)
To determine the cytokine mRNA expression levels, total RNA was isolated from LPS-treated RAW264.7 cells with TRIzol reagent (Gibco BRL), according to the manufacturer's instructions. Total RNA was stored at −70 °C until use. Analysis of mRNA was also performed using semiquantitative RT-PCR according to the manufacturer's instructions (Bioneer, Daejeon, Korea) as reported previously21. The results were expressed as the ratio of the optical density at 280 nm to the GAPDH mRNA concentration. The primers (Bioneer) used are listed in Table 1.
Luciferase reporter gene activity assay
HEK293 cells (1×106 cells/ml) were transfected with 1 μg of NF-κB-Luc, CREB-Luc, or AP-1-Luc plasmid, in addition to β-galactosidase plasmid, in the presence or absence of an Akt construct using the polyethyleneimine method in a 12-well plate according to the manufacturer's protocol22. The cells were used for experiments 48 h after transfection. Luciferase assays were performed using the Luciferase Assay System (Promega) as previously described23,24.
Preparation of total lysates and nuclear fractions, immunoblotting, and immunoprecipitation
RAW264.7 cells (5×106 cells/mL) or livers were washed 3 times in cold PBS with 1 mmol/L sodium orthovanadate and lysed in lysis buffer (20 mmol/L Tris-HCl, pH 7.4, 2 mmol/L EDTA, 2 mmol/L ethyleneglycotetraacetic acid, 50 mmol/L β-glycerophosphate, 1 mmol/L sodium orthovanadate, 1 mmol/L dithiothreitol, 1% Triton X-100, 10% glycerol, 10 μg/mL aprotinin, 10 μg/mL pepstatin, 1 mmol/L benzimide, and 2 mmol/L PMSF) for 30 min with rotation at 4 °C. The lysates were clarified by centrifugation at 16 000×g for 10 min at 4 °C and stored at -20 °C until needed.
Nuclear lysates were prepared in a three-step procedure25. After treatment, cells were collected with a rubber policeman, washed with 1×PBS, and lysed in 500 μL of lysis buffer on ice for 4 min. The cell lysates were then centrifuged at 19 326×g for 1 min in a microcentrifuge. In the second step, the pellet (the nuclear fraction) was washed once in wash buffer, which was the same as the lysis buffer without Nonidet P-40. In the final step, nuclei were treated with an extraction buffer containing 500 mmol/L KCl, 10% glycerol, and several other reagents as in the lysis buffer. The nuclei/extraction buffer mixture was frozen at -80 °C, thawed on ice and centrifuged at 19 326×g for 5 min. The supernatant was collected as the nuclear extract.
For immunoprecipitation, cell lysates containing equal amounts of protein (500 μg) from RAW264.7 cells cultured at 1×107 cells/mL and treated or not treated with LPS (1 μg/mL) for 2.5 min were pre-cleared with 10 μL of protein A-coupled Sepharose beads (50% v/v) (Amersham, UK) for 1 h at 4 °C. Pre-cleared samples were incubated with 5 μL of anti-Akt antibody overnight at 4 °C. Immune complexes were mixed with 10 μL of protein A-coupled Sepharose beads (50% vv) and stirred by rotation for 3 h at 4 °C.
Soluble cell lysates or boiled immunoprecipitated beads were analyzed on Western blots, and the phosphorylated or total transcription factors (p65, c-Jun, and c-Fos), MAPK proteins (ERK, p38, and JNK), IκBα, IKKα/β, Akt, p85/PI3K, PDK1, γ-tubulin, β-actin, and non-receptor tyrosine kinases (Src and Syk) were visualized as previously reported26.
Akt and PI3K kinase assays
To evaluate the Akt- and PI3K kinase-inhibitory activity in the extracts using purified enzymes, a kinase profiler service from Millipore (http://www.millipore.com/life_sciences/flx4/ld_kinases) was used. In a final reaction volume of 25 μL, Akt (1, 2, and 3) or PI3K (α, β, and γ; human; 1–5 mU) protein was incubated with the reaction buffer. The reaction was initiated by the addition of MgATP. After incubation for 40 min at room temperature, the reaction was stopped by adding 5 mL of a 3% phosphoric acid solution. Ten microliters of the reaction product was then spotted onto a GF/P30 filtermat (PerkinElmer, Inc), which was then washed three times for 5 min in 75 mmol/L phosphoric acid and once in methanol prior to drying and scintillation counting.
EtOH/HCl-induced gastritis, LPS-induced hepatitis, and acute toxicity tests
Inflammation of the stomach was induced with EtOH/HCl according to a published method27. Fasted ICR mice were orally treated with compound 7 (10 to 40 mg/kg) or ranitidine (40 mg/kg) suspended in 5% NaCMC. Thirty minutes later, 400 μL of 60% ethanol in 150 mmol/L HCl was administered orally. Each animal was euthanized with an overdose of urethane 1 h after the administration of the necrotizing agents. The stomach was excised and gently rinsed under running tap water. After opening the stomach along the greater curvature and spreading it out on a board, the area (mm2) of the mucosal erosive lesions was measured using a pixel-counter. Inflammation of the liver was induced by the injection of LPS according to a published method28. Fasted C57BL/6 mice were orally treated with compound 7 (20 mg/kg) once per day for 6 d. One hour after the final administration, LPS (10 mg/kg) was intraperitoneally administered. Each animal was anesthetized with an overdose of urethane 1 h after the administration of hepatitis inducers, and blood was drawn from the portal vein. The livers were then excised and gently rinsed under running tap water. Serum was obtained by centrifugation of the blood at 3000 r/min for 15 min. The levels of serum alanine aminotransferase (ALT) and aspartate aminotransferase (AST) were measured with a Roche Modular spectrophotometric autoanalyser. In the acute toxicity test, compound 7 was dissolved in 20% PEG 400 diluted in 5% BSA in H2O or suspended in 5% NaCMC and administered orally or by intraperitoneal injection. Lethality and body weight changes were determined after 7 d.
Statistical analysis
Data represent the mean±standard deviations (SD) from at least three independent experiments, each performed in triplicate, or are representative of three different experiments with similar results. For statistical comparisons, the results were analyzed using analysis of variance with Scheffe's post-hoc test and the Kruskal-Wallis/Mann-Whitney U-test. A P value <0.05 was considered to represent a significant difference. All statistical tests were carried out using SPSS (Statistical Package for the Social Sciences, SPSS Inc, Chicago, IL, USA).
Results and Discussion
In a search for novel anti-inflammatory drugs, we selected compounds (Figure 1A) similar in structure to BAY 11-7082, an IKK inhibitor, and tested them for inhibitory effects on the inflammatory mediators NO, TNF-α, and PGE2. Compound 7 [8-(tosylamino)quinoline] suppressed NO production in a dose-dependent manner without affecting cell viability (Figure 2), whereas the other compounds tested showed no inhibitory effect (Figure 1B). Compound 7 suppressed the release of NO, PGE2, and TNF-α by RAW264.7 cells (Figure 2A), peritoneal macrophages (Figure 2B), and bone-marrow derived macrophages (Figure 2C) during LPS exposure, with IC50 values of 1 to 5 μmol/L (Table 2). The inhibitory activity of compound 7 was comparable to that of BAY11-7082 (Figure 1C) when BAY11-7082 was tested as a treatment for arthritis11. These findings support the further development of compound 7 as an anti-inflammatory drug.

Effects of compounds 1 through 7 and BAY11-7082 on NO production. (A) Chemical structures of compounds 1 through 7. (B and C left panel) The Griess assay was used to determine the NO levels in culture supernatants of RAW264.7 cells or peritoneal macrophages treated with the test compounds and LPS (1 μg/mL) for 12 or 24 h. (C right panel) The viability of RAW264.7 cells was determined by the MTT assay. Mean±SD. n=4. bP<0.05, cP<0.01 vs the control.

Effect of compound 7 on the production of inflammatory mediators. (A, B and C) The levels of NO, PGE2, and TNF-α were determined by the Griess assay, EIA, and ELISA in culture supernatants of RAW264.7 cells, peritoneal macrophages or bone marrow-derived macrophages treated with compound 7 and LPS (1 μg/mL) for 6 or 24 h. The viability of RAW264.7 cells and peritoneal macrophages was determined using the MTT assay. bP<0.05, cP<0.01 vs the control.
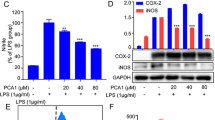
To explore the inhibitory mechanism of compound 7, we analyzed the effects of this compound on the transcription of genes encoding inflammatory mediators. Compound 7 dose-dependently reduced the levels of TNF-α, IL-1β, IL-6, IL-12, COX-2, and iNOS mRNAs, implying that this drug inhibits inflammatory mediator production at the level of transcription (Figure 3). To identify the transcription factors targeted by compound 7, we measured transcription factor levels in nuclear fractions in cells exposed to LPS as an inflammatory stimulus. In this setting, compound 7 suppressed p65 upregulation after 15 min and 2 h without exerting inhibitory effects on c-Jun and c-Fos (Figure 4A left panel). We found a similar pattern of inhibition in LPS-treated peritoneal macrophages (Figure 4A, right panel). In the reporter gene assay performed with constructs containing NF-κB, CREB or AP-1 binding promoters, similar inhibitory patterns for the activation of transcription factors were observed. Thus, compound 7 suppressed NF-κB-mediated luciferase activities that were stimulated by PMA or cotransfection with other adaptor molecules such as TRIF and MyD88 (Figure 4B), but this compound did not suppress AP-1 or CREB activities (Figure 4C). These results indicate that compound 7 modulates NF-κB signaling at an early stage.

Effect of compound 7 on the expression of pro-inflammatory genes. The levels of iNOS, COX-2, TNF-α, IL-1β, IL-6, and IL-12 p40 mRNAs were determined by semiquantitative RT-PCR.

Effect of compound 7 on the activation of transcription factors (NF-κB and AP-1). (A) The levels of NF-κB (p65), STAT-1, and AP-1 (c-Jun/c-Fos) in nuclear fractions from LPS-treated RAW264.7 cells and peritoneal macrophages were determined by immunoblotting analysis with antibodies against the total proteins. (B and C) HEK293 cells cotransfected with plasmid constructs for NF-κB-Luc, CREB-Luc or AP-1-Luc; adaptor molecules (MyD88 and TRIF) (each at 1 μg/mL); and β-gal (as a transfection control) were treated with compound 7, BAY (BAY11-7082) or U0 (U0126) in the presence or absence of PMA (100 nmol/L). The luciferase activity was measured using a luminometer. bP<0.05, cP<0.01 vs the control.
Because NF-κB activation is linked to a cascade of kinase activation, we sought to identify the exact target of compound 7 by determining the levels of phosphorylated IκBα, IKK, Akt, PDK1, PI3K, and Src or Syk after LPS stimulation. At 5 and 60 min, compound 7 strongly suppressed IκBα phosphorylation (Figure 5A). Because of the effect at 5 min, we turned our attention to early signaling events to investigate the target of compound 7. Interestingly, IκBα and its upstream kinase IKKα/β were phosphorylated at 1 min, and Akt phosphorylation had not diminished at this time. In agreement with this result, compound 7 did not suppress the phosphorylation of Syk or Src, tyrosine kinases that contribute to IκBα phosphorylation at 5 min29. These data imply that compound 7 targets molecules upstream of IKK, such as Akt kinase, either directly or indirectly. To confirm these possibilities, a direct kinase assay was performed with purified PI3K and Akt. Contrary to expectations, there was no inhibition of any type of PI3K and Akt (Figure 5C). Nonetheless, the molecular association between Akt and IKK observed after LPS treatment was clearly reduced by treatment with compound 7 (Figure 5D), suggesting that this compound seems to target to the binding event between Akt and its substrate protein IKK. Meanwhile, to confirm that compound 7 does not inhibit AP-1, we evaluated the levels of phosphorylated MAPK-related kinases (ERK, JNK, and p38). As expected, compound 7 did not suppress the phosphorylation of these enzymes (Figure 5E).

Effects of compound 7 on the activation of signaling enzymes upstream of NF-κB translocation. (A, B, and E) The levels of phospho- and total proteins of IκBα, IKKα/β, Akt, ERK, p38, JNK, and β-actin in cell lysates were determined using phospho-specific and total-protein antibodies, respectively. (C) The kinase activities of Akt and PI3K were determined by a direct kinase assay using purified enzymes. The control activity of each enzyme (Akt or PI3K) was set to 100%. (D) The effects of compound 7 on the formation of the signaling complex composed of Akt and phospho-IKK in the total lysates from LPS-treated RAW264.7 cells (5×106 cells/mL) were determined by immunoprecipitation with an anti-Akt antibody and immunoblotting with antibodies to p-IKK.
The Akt pathway, considered to be one of the major targets of compound 7, was initially recognized as a significant cell survival signal and subsequently as a potential target for cancer therapy. Based on recent evidence, however, attention has shifted to the role of Akt signaling in LPS-induced inflammatory responses30. It is noteworthy that anti-inflammatory herbal extracts from Eleutherococcus senticosus, Dichroa feb rifuga, and Cymbopogon citratus31,32,33 and ethnopharmacological agents such as curcumin, resveratrol, and quercetin34,35,36 may suppress the Akt pathway. The Akt inhibitors LY294002 and wortmannin displayed anti-inflammatory activity, reflected by the suppression of NO and PGE2 (Figure 6A). Furthermore, the overexpression of Akt induced NF-κB activation up to 2.5 fold as assessed by measuring NF-κB-mediated luciferase activity (Figure 6B). These findings suggest that Akt pathway inhibition may be a determining event in compound 7-mediated anti-inflammatory action. We will investigate how this compound mediates Akt-IKK binding inhibition without affecting Akt kinase activity in future experiments in our lab. In these experiments, the mode of binding between Akt and IKK and the inhibition of this binding by compound 7 will be analyzed using mutant constructs of these proteins and immunoprecipitation.

Functional role of Akt in the induction of inflammatory responses. (A) Culture supernatants prepared from LPS-treated RAW264.7 cells pre-treated with standard PI3K/Akt inhibitors [LY294002 (LY) and wortmannin (Wort)] were assayed for NO, TNF-α, and PGE2. (B) HEK293 cells were cotransfected with plasmid constructs for either NF-κB-Luc or Akt (1 μg/mL each) and β-gal (as a transfection control). The luciferase activity was measured with a luminometer. bP<0.05, cP<0.01 vs the control. eP<0.05, fP<0.01 vs the normal.
To determine whether compound 7 is active by the oral route, we tested its effect against EtOH/HCl-stimulated gastritis, an in vivo model of inflammatory disease that is widely used in drug development. At a single dose of 40 mg/kg, compound 7 significantly reduced gastric tissue injury following EtOH/HCl administration, as did ranitidine (40 mg/kg), the positive control (Figure 7A). In addition, this compound also suppressed LPS-induced hepatitis symptoms as assessed by measuring the serum levels of enzymes (ALT and AST) indicative of liver damage, implying that in vivo TLR4-mediated inflammatory symptoms are also ameliorated by this compound. The acute administration of compound 7 to mice at 500 mg/kg for 1 week by the oral or intraperitoneal route induced no perturbation in body weight or change in mortality (Figure 7C). These results support the further testing of compound 7 as an orally available, well-tolerated anti-inflammatory drug37.

Effects of compound 7 on gastric inflammation in mice induced by HCl/EtOH treatment and on acute toxicity. (A) Mice were treated orally with compound 7 or ranitidine for 3 d and then treated orally with HCl/EtOH. After 24 h, the gastric lesions in the stomach were measured by pixel counting right panel) and photographed (left panel). The area of gastric lesions present after treatment with the inducer alone is represented by 100%. (B) Mice orally treated with compound 7 for 6 d were intraperitoneally treated with LPS. After 1 h, serum was prepared to measure the biochemical parameters (AST and ALT) (upper panel), and the levels of p-IKK and β-actin were analyzed by Western blotting of the liver lysate (lower panel). (C) Lethality and body weight changes were followed for 7 d after the oral administration (1 g/kg) or intraperitoneal injection of compound 7 (500 mg/kg). bP<0.05 vs the control.
In summary, we have shown that compound 7 may suppress the production of NO, TNF-α, and PGE2 in LPS-treated macrophages and may attenuate HCl/EtOH-induced gastritis. In exploring the anti-inflammatory mechanism of compound 7, we found that this compound can block NF-κB activation by suppressing upstream signaling by IκBα, IKK, and Akt (Figure 8). We propose that compound 7 might be used as a novel anti-inflammatory drug. To investigate this application, we will test compound 7 for efficacy in vivo using models of acute disorders (septic shock and carrageenan-induced arthritis) and chronic disorders (collagen- or adjuvant-induced arthritis) in a pre-clinical study.

Putative pathway of the inhibition of LPS-activated inflammatory signaling by compound 7.
Author contribution
Yongwoo JUNG, Sungyoul HONG, and Jae Youl CHO designed the research; Yongwoo JUNG, Se Eun BYEON, Dae Sung YOO, Tao YU, Yanyan YANG, Ji Hye KIM, Eunji Kim, Yong Gyu LEE, and Deok JEONG performed the research; Yongwoo JUNG, Man Hee RHEE, Eui Su CHOUNG, Sungyoul HONG, and Jae Youl CHO analyzed the data; and Jae Youl CHO wrote the paper.
Abbreviations
ERK, extracellular signal-related kinase; TLR, Toll-like receptors; MAPK, mitogen-activated protein kinase; NF-κB, nuclear factor-κB; AP-1, activator protein-1; JNK, c-Jun N-terminal kinase; Akt, protein kinase B; ATF2, activating transcription factor 2; CREB, cAMP response element-binding; IKK, IκBα kinase; IRAK1, interleukin-1 receptor-associated kinase 1; MKK, MAP kinase kinase; MyD88, myeloid differentiation primary-response protein-88; TRAF6, tumor necrosis factor-receptor-associated factor-6; TAK1, TGF-β-activated kinase-1; PDK1, phosphoinositide-dependent protein kinase-1; Syk, spleen tyrosine kinase; TRIF, TIR-domain-containing adapter-inducing interferon-β; EIA, enzyme immunoassay; ELISA, enzyme-linked immunosorbent assay; MTT, 3-4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide; PI3K, phosphoinositide 3-kinase; LPS, lipopolysaccharide; RT-PCR, reverse transcriptase polymerase chain reaction; ALT, serum alanine aminotransferase; AST, aspartate aminotransferase
References
Kinne RW, Brauer R, Stuhlmuller B, Palombo-Kinne E, Burmester GR . Macrophages in rheumatoid arthritis. Arthritis Res 2000; 2: 189–202.
Owens T, Babcock AA, Millward JM, Toft-Hansen H . Cytokine and chemokine inter-regulation in the inflamed or injured CNS. Brain Res Brain Res Rev 2005; 48: 178–84.
Eizirik DL, Colli ML, Ortis F . The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol 2009; 5: 219–26.
Brown KD, Claudio E, Siebenlist U . The roles of the classical and alternative nuclear factor-kappa B pathways: potential implications for autoimmunity and rheumatoid arthritis. Arthritis Res Ther 2008; 10: 212.
Grieco FA, Vendrame F, Spagnuolo I, Dotta F . Innate immunity and the pathogenesis of type 1 diabetes. Semin Immunopathol 2011; 33: 57–66.
Tsan MF, Gao B . Endogenous ligands of Toll-like receptors. J Leukoc Biol 2004; 76: 514–9.
Mendes Sdos S, Candi A, Vansteenbrugge M, Pignon MR, Bult H, Boudjeltia KZ, et al. Microarray analyses of the effects of NF-kappaB or PI3K pathway inhibitors on the LPS-induced gene expression profile in RAW264.7 cells: synergistic effects of rapamycin on LPS-induced MMP9-overexpression. Cell Signal 2009; 21: 1109–22.
Min KJ, Lee JT, Joe EH, Kwon TK . An IkappaBalpha phosphorylation inhibitor induces heme oxygenase-1 (HO-1) expression through the activation of reactive oxygen species (ROS)-Nrf2-ARE signaling and ROS-PI3K/Akt signaling in an NF-kappaB-independent mechanism. Cell Signal 2011; 23: 1505–13.
Lee H, Lin CI, Liao JJ, Lee YW, Yang HY, Lee CY, et al. Lysophospholipids increase ICAM-1 expression in HUVEC through a Gi- and NF-kappaB-dependent mechanism. Am J Physiol Cell Physiol 2004; 287: C1657–66.
Dai Y, Rahmani M, Grant S . Proteasome inhibitors potentiate leukemic cell apoptosis induced by the cyclin-dependent kinase inhibitor flavopiridol through a SAPK/JNK- and NF-kappaB-dependent process. Oncogene 2003; 22: 7108–22.
Pierce JW, Schoenleber R, Jesmok G, Best J, Moore SA, Collins T, et al. Novel inhibitors of cytokine-induced IkappaBalpha phosphorylation and endothelial cell adhesion molecule expression show anti-inflammatory effects in vivo. J Biol Chem 1997; 272: 21096–103.
Yu T, Ahn HM, Shen T, Yoon K, Jang HJ, Lee YJ, et al. Anti-inflammatory activity of ethanol extract derived from Phaseolus angularis beans. J Ethnopharmacol 2011; 137: 1197–206.
Yu T, Lee YJ, Jang HJ, Kim AR, Hong S, Kim TW, et al. Anti-inflammatory activity of Sorbus commixta water extract and its molecular inhibitory mechanism. J Ethnopharmacol 2011; 134: 493–500.
Pyo S . The mechanism of poly I:C-induced antiviral activity in peritoneal macrophage. Arch Pharm Res 1994; 17: 93–9.
Jang SA, Kang SC, Sohn EH . Phagocytic effects of beta-glucans from the mushroom coriolus versicolor are related to Dectin-1, NOS, TNF-alpha signaling in macrophages. Biomol Ther 2011; 19: 438–44.
Cho JY, Baik KU, Jung JH . Park MH . In vitro anti-inflammatory effects of cynaropicrin, a sesquiterpene lactone, from Saussurea lappa. Eur J Pharmacol 2000; 398: 399–407.
Kang TJ, Moon JS, Lee S, Yirn D . Polyacetylene compound from Cirsium japonicum var ussuriense inhibits the LPS-Induced inflammatory reaction via suppression of NF-kappa B activity in RAW 264.7 cells. Biomol Ther 2011; 19: 97–101.
Jung YH, Park KY, Jeon JH, Kwak YS, Song YB, Wee JJ, et al. Red ginseng saponin fraction A isolated from Korean red ginseng by ultrafiltration on the porcine coronary artery. J Ginseng Res 2011; 35: 325–30.
Pauwels R, Balzarini J, Baba M, Snoeck R, Schols D, Herdewijn P, et al. Rapid and automated tetrazolium-based colorimetric assay for the detection of anti-HIV compounds. J Virol Methods 1988; 20: 309–21.
Kim JR, Oh DR, Cha MH, Pyo BS, Rhee JH, Choy HE, et al. Protective effect of polygoni cuspidati radix and emodin on Vibrio vulnificus cytotoxicity and infection. J Microbiol 2008; 46: 737–43.
Kim YJ, Lee JH, Lee OR, Shim JS, Jung SK, Son NR, et al. Isolation and characterization of a type II peroxiredoxin gene from Panax ginseng C. A. Meyer. J Ginseng Res 2010; 34: 296–303.
Jin M, Park S, Pyo MY . Suppressive effects of T-412, a flavone on interleukin-4 production in T cells. Biol Pharm Bull 2009; 32: 1875–9.
Jung KK, Lee HS, Cho JY, Shin WC, Rhee MH, Kim TG, et al. Inhibitory effect of curcumin on nitric oxide production from lipopolysaccharide-activated primary microglia. Life Sci 2006; 79: 2022–31.
Shen T, Lee J, Park MH, Lee YG, Rho HS, Kwak YS, et al. Ginsenoside Rp(1), a ginsenoside derivative, blocks promoter activation of iNOS and COX-2 genes by suppression of an IKK beta-mediated NF-kappa B pathway in HEK293 cells. J Ginseng Res 2011; 35: 200–8.
Byeon SE, Lee YG, Kim BH, Shen T, Lee SY, Park HJ, et al. Surfactin blocks NO production in lipopolysaccharide-activated macrophages by inhibiting NF-kappaB activation. J Microbiol Biotechnol 2008; 18: 1984–9.
Sohn EH, Jang SA, Lee CH, Jang KH, Kang SC, Park HJ, et al. Effects of Korean red ginseng extract for the treatment of atopic dermatitis-like skin lesions in mice. J Ginseng Res 2011; 35: 479–86.
Okabe S, Miyake H, Awane Y . Cytoprotective effects of NC-1300 and omeprazole on HCl. ethanol-induced gastric lesions in rats. Jpn J Pharmacol 1986; 42: 123–33.
Cho JY, Yeon JD, Kim JY, Yoo ES, Yu YH, Park MH . Hepatoprotection by human epidermal growth factor (hEGF) against experimental hepatitis induced by D-galactosamine (D-galN) or D-GalN/lipopolysaccharide. Biol Pharm Bull 2000; 23: 1243–6.
Lee YG, Chain BM, Cho JY . Distinct role of spleen tyrosine kinase in the early phosphorylation of inhibitor of kappaB alpha via activation of the phosphoinositide-3-kinase and Akt pathways. Int J Biochem Cell Biol 2009; 41: 811–21.
Lee YG, Lee J, Byeon SE, Yoo DS, Kim MH, Lee SY, et al. Functional role of Akt in macrophage-mediated innate immunity. Front Biosci 2011; 16: 517–30.
Jung CH, Jung H, Shin YC, Park JH, Jun CY, Kim HM, et al. Eleutherococcus senticosus extract attenuates LPS-induced iNOS expression through the inhibition of Akt and JNK pathways in murine macrophage. J Ethnopharmacol 2007; 113: 183–7.
Park SY, Park GY, Ko WS, Kim Y . Dichroa febrifuga Lour inhibits the production of IL-1beta and IL-6 through blocking NF-kappaB, MAPK and Akt activation in macrophages. J Ethnopharmacol 2009; 125: 246–51.
Francisco V, Figueirinha A, Neves BM, Garcia-Rodriguez C, Lopes MC, Cruz MT, et al. Cymbopogon citratus as source of new and safe anti-inflammatory drugs: bio-guided assay using lipopolysaccharide-stimulated macrophages. J Ethnopharmacol 2011; 133: 818–27.
Poolman TM, Ng LL, Farmer PB, Manson MM . Inhibition of the respiratory burst by resveratrol in human monocytes: correlation with inhibition of PI3K signaling. Free Radic Biol Med 2005; 39: 118–32.
Larmonier CB, Midura-Kiela MT, Ramalingam R, Laubitz D, Janikashvili N, Larmonier N, et al. Modulation of neutrophil motility by curcumin: implications for inflammatory bowel disease. Inflamm Bowel Dis 2011; 17: 503–15.
Ruiz PA, Braune A, Holzlwimmer G, Quintanilla-Fend L, Haller D . Quercetin inhibits TNF-induced NF-kappaB transcription factor recruitment to proinflammatory gene promoters in murine intestinal epithelial cells. J Nutr 2007; 137: 1208–15.
Byeon SE, Chung JY, Lee YG, Kim BH, Kim KH, Cho JY . In vitro and in vivo anti-inflammatory effects of taheebo, a water extract from the inner bark of Tabebuia avellanedae. J Ethnopharmacol 2008; 119: 145–52.
Acknowledgements
This work was supported by a National Research Foundation of Korea (NRF) grant funded by the Korean government (MEST) (to Jae Youl CHO) (No 2011-0016397).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Jung, Y., Byeon, S., Yoo, D. et al. 8-(Tosylamino)quinoline inhibits macrophage-mediated inflammation by suppressing NF-κB signaling. Acta Pharmacol Sin 33, 1037–1046 (2012). https://doi.org/10.1038/aps.2012.52
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2012.52
Keywords
This article is cited by
-
Therapeutic effects of DZ2002, a reversible SAHH inhibitor, on lupus-prone NZB×NZW F1 mice via interference with TLR-mediated APC response
Acta Pharmacologica Sinica (2014)
-
Synthesis and further rearrangements of 7-(2-cycloalken-1-yl)-8-quinolinols
Monatshefte für Chemie - Chemical Monthly (2014)
