
Abstract
Aim:
To construct a lentivirus-based inhibitor with specific secondary structure that could exert long-term suppression on microRNA-338-3p (miR-338-3p), thus elucidating its molecular function in colorectal carcinoma cells.
Methods:
The miR-338-3p inhibitor sequence was synthesized and inserted into pLV-THM plasmid. HEK-293T cells were co-transfected with the lentiviral vectors pLV-THM-miR-338-3p-inhibitor, psPAX2, and pMD2.G. The supernatant containing the lentivirus particles was harvested to determine the viral titer, and then used to infect colorectal carcinoma-derived SW-620 cells. eGFP(+) cells were sorted using flow cytometry. The expression of miR-338-3p in SW-620 cells was determined with real-time RT-PCR, and the expression of the smoothened (SMO) protein was detected using Western blot analysis. The migration ability of the transfected SW-620 cells was assessed with transwell assay.
Results:
Restriction endonuclease analysis and DNA sequencing demonstrated that the lentiviral vector pLV-THM-miR-338-3p-inhibitor was successfully constructed. The expression of miR-338-3p in SW-620 cells was significantly decreased by infection with the lentivirus pLV-THM-miR-338-3p-inhibitor. Moreover, the down-regulated expression of miR-338-3p caused up-regulated expression of the SMO protein in SW-620 cells, which showed significantly enhanced migration in transwell assay.
Conclusion:
The construction of the lentiviral vector pLV-THM-miR-338-3p-inhibitor with specific secondary structure provides a basis for further studies the molecular function of miR-338-3p in colorectal carcinoma. miR-338-3p may suppress SMO gene expression and thereby inhibit colorectal carcinoma migration.
Similar content being viewed by others


Introduction
MicroRNAs (miRNAs) are a new class of small, noncoding RNAs (18–25 nt) that regulate the expression of target genes through either translational repression or mRNA cleavage/decay1. miRNAs are known to play important roles in various biological processes, such as development, differentiation and cellular defense responses to infection. These nucleic acids have now been reported to be associated with many types of tumors2. Therefore, analysis of the underlying molecular mechanisms of miRNAs and tumor development is likely to become an important field of research. The development of reagents that can strongly suppress specific miRNAs has also generated much interest and will be important for both basic miRNA research and possible therapeutic strategies.
In recent studies, the majority of the synthetic, low-molecular weight inhibitors of specific miRNAs are based on anti-miRNA antisense oligonucleotides, in which some or all the ribonucleotides are modified to 2′-O-methylated RNA3, locked nucleic acids (LNA) or 2′-methoxyethylated RNA. These modifications provide resistance to cellular nucleases and increase the affinity of these oligonucleotides toward complementary miRNA sequences4. In addition, some antisense oligonucleotides have either flanking sequences or are connected to lipids via linkers5,6. However, because these reagents are diluted by successive cell divisions and in some cases are metabolized in the cytoplasm, their biological effects are often transient7. As a result, a continuous input of large quantities of dsRNA is required to sustain the down-regulation of miRNAs and RNAi. To achieve the long-term suppression of a specific miRNA, specialized plasmid- and viral-vectors carrying expression units for inhibitory RNA molecules have also been developed, including “antagomir”, “eraser” and “sponge”8.
miR-338-3p was a recently discovered miRNA and was found to be involved in cell differentiation. Although miR-338-3p is known to be specifically expressed in neuronal tissue, little is known about its abundance and function during the carcinogenesis of certain cancers9. We constructed a lentiviral vector, pLV-THM-miR-338-3p, and successfully over-expressed this vector in the colorectal carcinoma (CRC)-derived cell line SW-620 in our previous study. This provides the basis for further studies regarding the molecular function of miR-338-3p in CRC. For these studies, we utilized a decoy RNA system that uses a lentivirus-based vector containing RNA expression cassettes to enable the long-term suppression of miR-338-3p. The lentiviral vector is derived from HIV-I and is frequently used to deliver target genes because of its high transduction efficiency and stable integration of the genome into non-dividing cells10. The lentiviral vector is essential for the insertion of the object gene into the cell genome, and the regulatory efficiency of the RNA inhibitor depends on its structural design. Vermeulen et al11 demonstrated that an miRNA inhibitor containing 8 bp hairpin structures significantly suppressed miR-21 expression in HeLa cells. Furthermore, Haraguchi et al12 synthesized a series of RNA decoys with stem-loop structures and proved that the 3-nt linkers that are clipped between the miRNA binding sites (MBS) and the stem RNAs result in the strongest inhibitory effects. Therefore, in this study, we will design a miR-338-3p inhibitor that is flanked by sequences that form 8 bp hairpin structures in conjunction with an MBS with a 3-nt linker on both sides. We will design an inhibitor with a combination of these two structures to establish a novel and potent miR-338-3p inhibitor based on a lentiviral vector. Furthermore, this inhibitor should have long-term effects within the cell.
Materials and methods
Design of the miRNA inhibitor sequence
The sequence of the mature miR-338-3p (5′-UCCAGCAUCAGUGAUUUUGUUG-3′) was obtained from miRBase (http://www.mirbase.org/). The lentiviral vectors used in this study are pLV-THM, psPAX2 and pMD2.G, which are a transfer vector, packaging plasmid and envelope plasmid, respectively. The vector information can be found on the Addgene website (http://www.addgene.org/). The inhibitor sequence was inserted into the transfer vector between the Mlu I and Cla I restriction sites according to the Addgene protocol.
The MBS was then flanked by 3-nt linkers, which include an arbitrary sequence and 8 bp hairpin structures. Because the initial sequence of the human gene is located approximately 30 nt downstream of the TATA box13, more than 4 bp of arbitrary nucleic acids were required in the 5′ flanking region upstream the 5′ hairpin structures. This was necessary to construct the vector in accordance with the cloning strategy outlined on the LentiWeb (http://lentiweb.com/cloning_strategies.php). The two restriction sites and their corresponding protective bases were generated at the ends of both the 5′ and 3′ flanking regions (Figure 1).

Design of miR-338-3p inhibitor sequence. (A) Representative structure of miR-338-3p inhibitor. (B) Schematic representation of the generation of miR-338-3p inhibitor expression cassettes driven by H1 promoter. Section 1, 3, 11, 13 are protective bases. Section 2 and 12 are restriction sites, Mlu I and Cla I respectively. Section 4 is the arbitrary nucleic acids. Section 5 and 9 are hairpin structures. Section 6 and 8 are 3-nt linkers. Section 7 is MBS. Section 10 is terminator (poly T). 81 mer synthetic oligonucleotides pairs are annealed and cloned between the Mlu I and Cla I sites to generate RNA inhibitor.
To gain a better understand of the secondary structure of the decoy RNA and to determine whether self-annealing could occur inside the MBS sequence, which would result in the decline of its affinity to miR-338-3p, we sent the sequence of our interested gene to the Vienna RNA WebServers (http://rna.tbi.univie.ac.at/) for thermodynamic ensemble prediction of our RNA inhibitor (Figure 2).

Design of secondary structure of decoy RNA. (A) Description of the detail in RNA inhibitor prediction. (B) Image of the secondary structure of RNA inhibitor.
Transfer vector construction
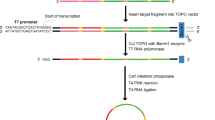
The third generation of self-inactivating, lentivirus plasmid, pLV-THM (HIV-1-based vector, Addgene, Cambridge, MA, USA), which contains a CMV-driven enhanced green fluorescence protein (eGFP) reporter and an H1 promoter upstream of the restriction sites (Mlu I and Cla I) to allow for the introduction of a short-hairpin RNA (shRNA) expression cassette, was used as the transfer plasmid and was linearized by digesting the vector with the restriction enzymes. The miR-338-3p inhibitor oligonucleotides described above were chemically synthesized by Sangon Biotech Co Ltd (Shanghai, China) and were inserted between the Mlu I and Cla I sites of the pLV-THM plasmid. After the miR-338-3p-inhibitor lentiviral-based vector was transformed into competent E coli DH5α cells using the calcium chloride method, antibiotic-resistant colonies were selected on LB-ampicillin agar plates. After colony selection and further propagation, the plasmid was extracted using the alkaline lysis method. The plasmid DNA was then analyzed by restriction enzyme digests and sequence analysis. The plasmid containing the target gene was digested with the restriction enzymes and amplified by PCR. The clones with positive PCR results were subjected to DNA sequencing. After synthesis of the oligonucleotide pairs that code for the decoy RNA that suppresses the endogenous miR-338-3p in cells, we cloned the oligonucleotide pairs between the Mlu I and Cla I sites of the plasmid, pLV-THM, to generate the RNA inhibitor (Figure 3).

The sketch of our cloning strategy of the transfer vector.
Cell lines and culture
Human embryonic kidney 293T (HEK-293T) cells (Invitrogen, Carlsbad, CA, USA) and the human CRC-derived cell line SW-620 (Shanghai Institutes for Biological Sciences) were cultured in Dulbecco's modified Eagle medium high glucose (H-DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Hyclone, USA) at 37 °C in a humidified incubator with 5% CO2. The medium was changed every 3 d, and the cells were trypsinized with trypsin/edetic acid when 80%–90% confluence was reached. Cells at passages 4–8 were used for the experiments.
Lentiviral packaging and virus collection
Twenty-four hours prior to transfection, the HEK-293T cells in logarithmic growth phase were trypsinized, and the cell density was adjusted to 1.0×106 cells/mL with complete culture medium. The cells were reseeded into 15-cm cell culture dishes and cultured for 24 h prior to transfection. The cells were 90%–95% confluent on the day of transfection. The recombinant viral vector encoding the miR-338-3p inhibitor and the two packaging plasmids psPAX2 and pMD2.G were extracted with a plasmid extraction kit (Invitrogen, Carlsbad, CA, USA) and co-transfected into HEK-293T cells according to the manufacturer's instructions. After 8 h of transfection, the cell culture medium was replaced with fresh complete medium. After 24 h of transfection, the expression of eGFP was determined. After 48 h of transfection, the culture medium was collected and centrifuged at 4000×g at 4 °C for 10 min to remove any cellular debris. The supernatant was filtered through a 0.45-μm filter into a Plus-20 centrifugal ultrafiltration unit and centrifuged at 4000×g to obtain a high-titer lentivirus stock. The lentivirus without the transgene was used as the negative control and was produced in the same manner as the inhibitor vector.
Virus condensation and titer assay
The high-titer lentivirus was used as the stock virus solution. HEK-293T cells were seeded in 96-well plates (4×104 cells/well in a volume of 100 μL) and cultured for 24 h. The medium was removed and 100 μL of the original virus solution and consecutive diluted virus solutions at dilutions of 10−1, 10−2, 10−3, 10−4, and 10−5 were added to the cells. After 48 h, 100 μL of complete medium was added to each well and the viral titer was determined by flow cytometry. The viral titer was calculated with the following formula: Titer (293T-transducing units/mL)=100 000 (target cells)×(% of eGFP-positive cells/100)/volume of supernatant in mL.
Virus transduction and fluorescent cell selection
SW-620 cells were seeded at 1.0×105 cells per well in 24-well plates in DMEM containing 10% FBS. After 24 h incubation, the cells were transduced with each lentivirus stock (3.0×105 Titer Unit). The SW-620 cells were then incubated for an additional 48–72 h prior to identifying the eGFP(+) cells by flow cytometry (Becton Dickinson, CA, USA).
Detection of miR-338-3p expression with real-time RT-PCR
Total RNA from SW-620 cells infected with the miR-338-3p expression vector was prepared using the TRIzol reagent (Invitrogen, Carlsbad, CA, USA) after viral transduction. The precipitate was dissolved in DEPC-treated water, and a nucleic acid protein analyzer (Beckman Coulter, USA) was used to determine the RNA concentration. The purity and integrity of the RNA were identified as follows: the A260 nm/A280 nm was ≥1.8, and the band ratio of 28S RNA to 18S RNA was ≥1.5 in formaldehyde denaturing gel electrophoresis. Accurate quantitation of the mature miR-338-3p was obtained using the TaqMan microRNA Assays (Applied Biosystems, Foster City, CA, USA). Briefly, the reverse transcription reaction was performed using 10 ng of total RNA and the looped primers. Real-time PCR was performed using the standard TaqMan microRNA Assays protocol on the iCycler iQ Real-Time PCR Detection System (Bio-Rad). The PCR reaction (20 μL) included 1.33 μL reverse transcription product, 1×TaqMan Universal PCR Master Mix, No AmpErase UNG, 0.2 μmol/L TaqMan probe, 1.5 μmol/L forward primer and 0.7 μmol/L reverse primer. The reactions were incubated in a 96-well plate at 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1 min. The miR-338-3p expression level was measured using the Ct (threshold cycle) method. The Ct is the fractional cycle number at which the fluorescence of each sample passes the fixed threshold. The ΔΔCt method for relative quantitation of gene expression was used to determine the miR-338-3p expression levels. The ΔCt was calculated by subtracting the Ct of U6 from the Ct of the miR-338-3p. The ΔΔCt was calculated by subtracting the ΔCt of the reference sample from the ΔCt of each sample. The fold change was calculated using the equation 2−ΔΔCt14. The Taqman microRNA Assays for U6 RNA was used to normalize the relative abundance of miR-338-3p. The above experiment was repeated at least three times.
miRNA target prediction
The analysis of miR-338-3p-predicted targets was performed using the algorithms TargetScan (http://targetscan.org/), PicTar (http://pictar.mdc-berlin.de/) and miRanda (http://www.microrna.org/microrna/home.do).
Detection of smoothened (SMO) protein expression by Western blot analysis
SW-620 cells were rinsed twice with cold PBS buffer and were then lysed in ice-cold lysis buffer containing 150 mmol/L NaCl, 50 mmol/L Tris-HCl (pH 7.6), 0.1% SDS, 1% Nonidet P-40, and protease inhibitor cocktail (Boehringer Mannheim, Lewes, UK). The samples were cleared by centrifugation at 13 000×g for 10 min. The cellular protein (50 μg) was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and electrotransferred to polyvinylidine fluoride (PVDF) membranes (Immobilon, Bedford, MA, USA). After blocking in 20 mmol/L Tris-HCl, (pH 7.6) containing 150 mmol/L NaCl, 0.1% Tween-20, and 5% non-fat dry milk, the membranes were incubated with primary antibodies against SMO or β-actin, which was used as a sample loading control, overnight at 4 °C. The membranes were then incubated with horseradish peroxidase-conjugated secondary antibody. The blot was developed using the ECL detection kit (Amersham Pharmacia Biotech) according to the manufacturer's instructions. This experiment was repeated at least three times.
Transwell migration assay
The migration assay was performed using transwell cell culture chambers (8 μmol/L pore size polycarbonate membrane, Costar). SW-620 cells were resuspended in DMEM with 0.1% FBS at a concentration of 3x105 cells/mL. The upper chamber was loaded with 100 μL cell suspension and the lower chamber was loaded with 600 μL DMEM supplemented with 10% FBS. After incubation for 48 h at 37 °C in a 5% CO2 atmosphere, the filter was fixed with 4% paraformaldehyde and stained with crystal violet. The cells on the upper side of the filter were wiped off with a cotton swab. The cells that migrated to the underside of the membrane were counted under a microscope. Ten microscopic fields (×400) were randomly selected for cell counting. This experiment was repeated at least three times.
Statistical analysis
The relative expression analysis of the target gene was performed using REST-XL (Relative Expression Software Tool, available at http://www.gene-quantification.info/). All data in the experiment were presented as the average±standard deviation (mean±SD). Comparisons between the groups were analyzed with a one-way ANOVA and Student-Newman-Keuls q test using the SPSS 13.0 software (SPSS Inc, Chicago, IL, USA). P values less than 0.05 were considered statistically significant.
Results
Analysis of the miR-338-3p-inhibitor sequence
As shown in Figure 2B, the MBS was unlikely to form a self-annealing sequence, and the other elements were not predicted to block the MBS. Therefore, the structure of the miRNA inhibitor should be completely complementary to the target miRNA sequence.
Packaging of the pLV-THM-miR-338-3p-inhibitor lentivirus and titer assay
HEK-293T cells were co-transfected with the transfer plasmid, pLV-THM-transgene, the packaging plasmid, psPAX2, and the envelope plasmid, pMD2.G. GFP was detected 24 h after co-transfection (Figure 4). Moreover, after 48 h infection with the diluted lentivirus, different levels of eGFP were also expressed in the HEK-293T cells (Figure 5). The viral titer was calculated as the proportion of fluorescent cells that was determined by flow cytometry, and the viral titer was 4.8×108 TU/mL.

HEK-293T cells transfected with recombinant plasmid, psPAX2 and pMD2.G. (A) HEK-293T cells under light microscope (20×); (B) HEK-293T cells under fluorescent microscope (20×).

HEK-293T cells transduced by lentivirus diluted in different density of 100, 10−1, 10−2, 10−3, 10−4, and 10−5. (A) HEK-293T cells under light microscope (20×); (B) HEK-293T cells under fluorescent microscope (20×).
Virus transduction and fluorescent cell identification
GFP was expressed 48 h after the SW-620 cells were transduced by the lentivirus, and the cells were observed under a fluorescence microscope (Figure 6A–6B). This suggests that the miR-338-3p-inhibitor vector was successfully transduced into the SW-620 cells. The eGFP(+) fluorescent cells were then identified using flow cytometry (Figure 6C–6E).

SW-620 cells infected by lentivirus (3.0×105 TU) before and after flow cytometry selection. (A, B) SW-620 cells infected by lentivirus before flow cytometry selection (A: light microscope; B: fluorescent microscope). (C) SW-620 cells with eGFP(+) were distinguished by flow cytometry. (D, E) SW-620 cells infected by lentivirus after flow cytometry selection (D: light microscope; E: fluorescent microscope).
Real-time RT-PCR for detection of miR-338-3p expression in a CRC-derived cell line
To study the expression pattern of miR-338-3p in SW-620 cells after lentivirus transduction, we performed real-time RT-PCR to detect miR-338-3p expression in the SW-620 cells. The real-time RT-PCR results indicated that the miR-338-3p cDNA increased exponentially and then plateaued. The miR-338-3p amplification curve was a typical reverse S pattern (Figure 7A) and showed higher amplification efficiency. The miR-338-3p PCR product was 72 bp long, the corresponding Tm was 84.09±0.15 °C, the melting temperature was even, and the shape of the peak was sharp (Figure 7B). As shown in Figure 7C, the expression level of miR-338-3p in the treatment group is less than one third of the expression in the control cells that were not transduced with the inhibitor vector.

Real-time RT-PCR analysis detecting miR-338-3p expression in SW-620 cells. (A) miR-338-3p cDNA concentrations' Log value as ordinate, Ct value as abscissa. (B) Tm of miR-338-3p was 84.09 °C. (C) Expression of miR-338-3p detected by real-time RT-PCR. Expression of U6 snRNA was used as internal control. bP<0.01 versus control group.
Biological functions of miR-338-3p in a CRC-derived cell line
To further investigate the functions of miR-338-3p in CRC cells, we constructed a SW-620 cell line with down-regulated miR-338-3p expression. This SW-620 cell line stably expressed the miR-338-3p inhibitor from the lentivirus vector, pLV-THM-miR-338-3p-inhibitor. Most miRs are hypothesized to control gene expression by base-pairing with the miR-recognizing elements (miR-RE) found in their messenger targets. We utilized all three currently available major prediction programs, TargetScan, miRanda and PicTar, to analyze the potential interactions between miR-338-3p and SMO. At least two of these algorithms (TargetScan and miRanda) reveal a potential miR-338-3p target site in the SMO mRNA 3′-UTR region (Figure 8A).

The biological functions of miR-338-3p on CRC-derived cell line. (A) SMO 3′-UTR site potentially targeted by miR-338-3p as predicted by TargetScan. (B) Western-blot analysis showing SMO protein expression in SW-620 cells. β-Actin was used as house-keeping gene to normalize SMO protein expression. Note: lane 1, Negative control; lane 2, SW-620 cells transfected with pLV-THM-control; lanes 3, SW-620 cells transfected with pLV-THM-miR-338-3p-inhibitor. The results are representative of three independent experiments. (C–E) Transwell assay showing migration ability of the blank control SW-620 cells (C), SW-620 cells transfected with pLV-THM-control (D) and SW-620 cells transfected with pLV-THM-miR-338-3p-inhibitor (E). Migrated cells found on the bottom side of the membrane were fixed and stained. Numbers of migrated cells on the membrane bottom were counted and shown at the top of each panel. Down-expression of miR-338-3p increased the expression of SMO protein in SW-620 cells, which showed obviously enhanced invasive ability in transwell assay. n=3. Mean±SD. bP<0.01 versus control group.
To determine whether miR-338-3p affects SMO expression in CRC cells, we analyzed the effects of miR-338-3p ectopic expression. We transduced the lentivirus vector, pLV-THM-miR-338-3p-inhibitor, into SW-620 cells and used Western blot analysis to determine whether there were changes in the SMO protein level. Introduction of the miR-338-3p-inhibitor caused a significant decrease in the level of miR-338-3p and increased the level of the SMO protein (Figure 8B). This result strongly suggests the post-transcriptional regulation of the SMO protein by miR-338-3p.
To gain a better understanding of changes in the cell phenotype, we used a transwell migration assay to examine the mobility of SW-620 cells transduced with the miR-338-3p-inhibitor. The results of this study demonstrated that a greater number of miR-338-3p-inhibitor-positive cells migrated to the lower side of the membrane than the miR-338-3p-inhibitor-negative cells (Figure 8C–8E).
Discussion
miRNAs play a key role in the regulation of basic biological activities, such as the growth, development, and differentiation of organisms, and because of this, unbalanced miRNA expression may contribute to many human diseases, particularly the occurrence and development of tumors14. miR-338-3p was a recently discovered miRNA that was found to be involved in cellular differentiation. It was reported that miR-338-3p was a tumor suppressor gene in central nervous system tumors, the expression of which was decreased in central nervous system tumor cell lines. Furthermore, the miR-338-3p sequence is located within intron-8 of the apoptosis-associated tyrosine kinase (AATK) gene, and optimal neurite growth requires not only the enzymatic activity of AATK but also the intronic miR-338-3p generated from the chromosomal AATK gene15. Tsuchiya et al16 have reported that miR-338-3p contributes to the formation of epithelial basolateral polarity by facilitating the translocalization of β1-integrin to the basolateral membrane, which highlights a potentially important role for miR-338-3p in the process of epithelial cell differentiation. Huang et al17 demonstrated that a decrease of miR-338-3p expression in hepatocellular carcinoma (HCC), which is another type of epithelial cell-derived cancer, was significantly associated with TNM stage, vascular invasion, intrahepatic metastasis, tumor size and tumor grade. In our previous study, we demonstrated that miR-338-3p expression was down-regulated in both CRC samples and CRC-derived cell lines. We also demonstrated that miR-338-3p was associated with an unfavorable disease-free survival that was independent of clinical factors in specimens from CRC patients treated by both curative surgery and adjuvant chemotherapy (data not shown). To confirm the molecular mechanism of miR-338-3p in CRC, it is necessary to observe the biological effects of the up- and down-regulation of miR-338-3p. We constructed a CRC cell line in which miR-338-3p was over-expressed. This cell line was constructed by transducing the lentivirus vector, pLV-THM-miR-338-3p, into SW-620 cells. However, the long-term suppression of miR-338-3p in CRC cells remains a problem that has yet to be solved.
For the stable expression of genes in target cells, lentivirus-based drugs or gene therapy is a potential therapeutic approach for the treatment of cancers. In recent years, lentiviral vectors have emerged as promising tools for anti-tumor immunotherapy and gene therapy because of their ability to transduce a wide variety of dividing and non-dividing cell types, including tumor cells, dendritic cells and T cells. The modification of cancer cells with various types of miRNAs that have been shown to be involved in tumor development is performed using lentiviral vectors in the majority of relative studies because of efficient transgene delivery that results in either the over-expression or knockdown of miRNAs both in vitro and in vivo18. These approaches have major implications for the adoptive cellular therapy of virus-associated malignancies and cancer19. In conclusion, the potential of lentiviral vectors as a generally applicable “off-the-shelf” therapeutic for anti-tumor biotherapy should be highlighted.
In this study, we successfully developed a novel lentivirus-based miR-338-3p inhibitor with a hairpin structure. Several previous studies have reported the construction of a RNA decoy with two MBS inside the stem-loop structure20. However, this structure may cause self-annealing between the two MBS when they are transcribed into RNA. This will most likely reduce the inhibitory potential of the RNA decoy21. Modifying the sequences of two MBS to prevent them from being complementary to each other is a sophisticated process. We chose the inhibitory structure of one MBS with two 8 bp hairpins flanking it on both sides and 3-nt linkers connecting the hairpin and MBS. Notably, we show that our RNA decoy can specifically inhibit endogenous miR-338-3p to induce the corresponding biological effect. In our study, we explored the relationship between the expression level of miR-338-3p and the specific biological effects in CRC, such as invasion and metastasis. The success of the miR-338-3p inhibitor is extremely beneficial for further research.
Recently, SMO, which is the key activator of the Hedgehog (Hh) signaling pathway, was reported to induce CRC invasion and metastasis by targeting the glioma-associated oncogene homologue 1 (GLI-1), a downstream target of the Hh signal22. With the application of bioinformatics prediction programs, such as TargetScan, PicTar and miRanda, we found that miR-338-3p and the 3′-UTR of SMO mRNA had complementary binding sites. From this, we hypothesized that SMO may be a new target of miR-338-3p in CRC; however, this finding has not yet been reported. In this study, the SMO protein was up-regulated in SW-620 cells transduced with pLV-THM-miR-338-3p-inhibitor, and this was confirmed by Western blot analysis. Moreover, decreased expression of miR-338-3p increased the expression of the SMO protein in SW-620 cells, which showed significantly enhanced invasive ability in a transwell assay. Consistent with Huang et al23, our data indicate that miR-338-3p suppresses SMO gene expression to inhibit CRC migration, which suggests that SMO is a potential anti-tumor candidate for the treatment and prevention of CRC.
Therefore, this tool could be applied to the analysis of regulatory networks that are formed either transiently or stably between miR-338-3p and coding genes. This is also applicable to the construction of a miR-338-3p knockdown mouse. Because the mouse U6 promoter regulated by Cre-loxP has already been well-established, time- and tissue-specific miRNA knockdown in the mouse should be possible using modified lentivirus vector expression systems for further studies regarding the alterations of biological functions in CRC24. Because of these properties, our miR-338-3p inhibitor will be very useful for future studies and may also have potential as a novel human gene therapy. Using recent evidence from pharmaceutical studies focusing on siRNA, our objective is to develop an efficient drug delivery system for a miRNA inhibitor. Furthermore, the reduction in potential side effects of the molecule when the miRNA is transduced by a lentivirus in vivo, such as unwanted cytokine responses, remains an additional goal.
Author contribution
Dr Kai SUN and Chen GUO designed the study, performed the study, analyzed the data, wrote the manuscript and revised the paper. Hai-jun DENG, Jing-qing DONG, Shang-tong LEI, and Guo-xin LI helped perform a portion of the study.
Abbreviations
CRC, colorectal carcinoma; miR, microRNA; 3′-UTR, 3′-untranslated region; MBS, miRNA binding site; SMO, smoothened
References
Bartels CL, Tsongalis GJ . MicroRNAs: novel biomarkers for human cancer. Clin Chem 2009; 55: 623–31.
Mirnezami AH, Pickard K, Zhang L, Primrose JN, Packham G . MicroRNAs: key players in carcinogenesis and novel therapeutic targets. Eur J Surg Oncol 2009; 35: 339–47.
Torres AG, Fabani MM, Vigorito E, Williams D, Al-Obaidi N, Wojciechowski F, et al. Chemical structure requirements and cellular targeting of microRNA-122 by peptide nucleic acids anti-miRs. Nucleic Acids Res 2012; 40: 2152–67.
Ebert MS, Sharp PA . MicroRNA sponges: progress and possibilities. RNA 2010; 16: 2043–50.
Ebert MS, Neilson JR, Sharp PA . MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat Methods 2007; 4: 721–6.
Ebert MS, Sharp PA . Emerging roles for natural microRNA sponges. Curr Biol 2010; 20: 858–61.
Stegmeier F, Hu G, Rickles RJ, Hannon GJ, Elledge SJ . A lentiviral microRNA-based system for single-copy polymerase II-regulated RNA interference in mammalian cells. Proc Natl Acad Sci U S A 2005; 102: 13212–7.
Reichel M, Li J, Millar AA . Silencing the silencer: strategies to inhibit microRNA activity. Biotechnol Lett 2011; 33: 1285–92.
Aschrafi A, Schwechter AD, Mameza MG, Natera-Naranjo O, Gioio AE, Kaplan BB . MicroRNA-338 regulates local cytochrome c oxidase IV mRNA levels and oxidative phosphorylation in the axons of sympathetic neurons. J Neurosci 2008; 28: 12581–90.
Park F . Lentiviral vectors: are they the future of animal transgenesis? Physiol Genomics 2007; 31: 159–73.
Vermeulen A, Robertson B, Dalby AB, Marshall WS, Karpilow J, Leake D, et al. Double-stranded regions are essential design components of potent inhibitors of RISC function. RNA 2007; 13: 723–30.
Haraguchi T, Nakano H, Tagawa T, Ohki T, Ueno Y, Yoshida T, et al. A potent 2′-O-methylated RNA-based microRNA inhibitor with unique secondary structures. Nucleic Acids Res 2012; 40: e58.
Tokusumi Y, Ma Y, Song X, Jacobson RH, Takada S . The new core promoter element XCPE1 (X Core Promoter Element 1) directs activator-, mediator-, and TATA-binding protein-dependent but TFIID-independent RNA polymerase II transcription from TATA-less promoters. Mol Cell Biol 2007; 27: 1844–58.
Sun K, Zeng JJ, Wang W, Wu CT, Lei ST, Li GX . MicroRNA-221 inhibits CDKN1C/p57 expression in human colorectal carcinoma. Acta Pharmacol Sin 2011; 32: 375–84.
Rodriguez A, Griffiths-Jones S, Ashurst JL, Bradley A . Identification of mammalian microRNA host genes and transcription units. Genome Res 2004; 14: 1902–10.
Tsuchiya S, Oku M, Imanaka Y, Kunimoto R, Okuno Y, Terasawa K, et al. MicroRNA-338-3p and microRNA-451 contribute to the formation of basolateral polarity in epithelial cells. Nucleic Acids Res 2009; 37: 3821–7.
Huang XH, Wang Q, Chen JS, Fu XH, Chen XL, Chen LZ, et al. Bead-based microarray analysis of microRNA expression in hepatocellular carcinoma: miR-338 is down-regulated. Hepatology Research 2009; 39: 786–94.
Hu B, Tai A, Wang P . Immunization delivered by lentiviral vectors for cancer and infectious diseases. Immunol Rev 2011; 239: 45–61.
Yue J . miRNA and vascular cell movement. Adv Drug Deliv Rev 2011; 63: 616–22.
Haraguchi T, Ozaki Y, Iba H . Vectors expressing efficient RNA decoys achieve the long-term suppression of specific microRNA activity in mammalian cells. Nucleic Acids Res 2009; 37: e43.
Scherr M, Venturini L, Battmer K, Schaller-Schoenitz M, Schaefer D, Dallmann I, et al. Lentivirus-mediated antagomir expression for specific inhibition of miRNA function. Nucleic Acids Res 2007; 35: e149.
Wang K, Pan L, Che X, Cui D, Li C . Sonic Hedgehog/GLI1 signaling pathway inhibition restricts cell migration and invasion in human gliomas. Neurol Res 2010; 32: 975–80.
Huang XH, Chen JS, Wang Q, Chen XL, Wen L, Chen LZ, et al. miR-338-3p suppresses invasion of liver cancer cell by targeting smoothened. J Pathol 2011; 225: 463–72.
Ventura A, Meissner A, Dillon CP, McManus M, Sharp PA, Van Parijs L, et al. Cre-lox-regulated conditional RNA interference from transgenes. Proc Natl Acad Sci U S A 2004; 101: 10380–5.
Acknowledgements
This research is supported by a grant from the National Natural Science Foundation of China (No 81101896).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sun, K., Guo, C., Deng, Hj. et al. Construction of lentivirus-based inhibitor of hsa-microRNA-338-3p with specific secondary structure. Acta Pharmacol Sin 34, 167–175 (2013). https://doi.org/10.1038/aps.2012.172
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2012.172
