
Abstract
Aim:
To investigate the effects of the potassium-sparing diuretic amiloride on endothelial cell apoptosis during lipopolysaccharide (LPS)-accelerated atherosclerosis.
Methods:
Human umbilical vein endothelial cells (HUVECs) were exposed to LPS (100 ng/mL) in the presence of drugs tested. The activity of Na+/H+ exchanger 1 (NHE1) and calpain, intracellular free Ca2+level ([Ca2+]i), as well as the expression of apoptosis-related proteins in the cells were measured. For in vivo study, ApoE-deficient (ApoE−/−) mice were fed high-fat diets with 0.5% (w/w) amiloride for 4 weeks and LPS (10 μg/mouse) infusion into caudal veins. Afterwards, atherosclerotic lesions, NHE1 activity and Bcl-2 expression in the aortic tissues were evaluated.
Results:
LPS treatment increased NHE1 activity and [Ca2+]i in HUVECs in a time-dependent manner, which was associated with increased activity of the Ca2+-dependent protease calpain. Amiloride (1−10 μmol/L) significantly suppressed LPS-induced increases in NHE1 activity, [Ca2+]i. and calpain activity. In the presence of the Ca2+ chelator BAPTA (0.5 mmol/L), LPS-induced increase of calpain activity was also abolished. In LPS-treated HUVECs, the expression of Bcl-2 protein was significantly decreased without altering its mRNA level. In the presence of amiloride (10 μmol/L) or the calpain inhibitor ZLLal (50 μmol/L), the down-regulation of Bcl-2 protein by LPS was blocked. LPS treatment did not alter the expression of Bax and Bak proteins in HUVECs. In the presence of amiloride, BAPTA or ZLLal, LPS-induced HUVEC apoptosis was significantly attenuated. In ApoE−/− mice, administration of amiloride significantly suppressed LPS-accelerated atherosclerosis and LPS-induced increase of NHE1 activity, and reversed LPS-induced down-regulation of Bcl-2 expression.
Conclusion:
LPS stimulates NHE1 activity, increases [Ca2+]i, and activates calpain, which leads to endothelial cell apoptosis related to decreased Bcl-2 expression. Amiloride inhibits NHE1 activity, thus attenuates LPS-accelerated atherosclerosis in mice.
Similar content being viewed by others

Introduction
Atherosclerosis is a common cardiovascular disease characterized by the deposition of fatty substances on and fibrosis of the inner lining of the arteries. Amiloride is a potassium-sparing diuretic used in the management of hypertension and congestive heart failure. Amiloride was also tested as a treatment of cystic fibrosis and an inhibitor of Na+/H+ exchanger 1 (NHE1)1. However, it has not been reported whether amiloride has a potentially beneficial effect as an anti-atherosclerotic treatment.
Lipopolysaccharide (LPS) is a component of the outer membrane of Gram-negative bacteria and elicits inflammatory responses in immune and non-immune cells, including endothelial cells2. LPS can induce endothelial cell injury by activating calpain3 and accelerate atherosclerosis in mice. During septic shock, endothelial cells are injured by LPS, which leads to vascular complications including thrombosis4,5. Indeed, systemic organ failure induced by LPS is instigated by vascular endothelial damage, including cell death, perivascular accumulation of leukocytes, and vascular occlusion6. Vascular endothelial apoptosis is mediated by various mechanisms, such as free radicals, cytokines, cell interaction and death receptors, all of which can contribute to atherosclerosis.
The induction of apoptosis by NHE1 activation has been extensively studied. NHE1 is ubiquitously expressed in the plasma membrane of eukaryotes and exchanges extracellular Na+ for intracellular H+ to regulate intracellular pH (pHi) value and Na+i concentration7. The activation of NHE1 increases Na+i concentration, which leads to an overload of [Ca2+]i through the Na+/Ca2+ exchanger that contributes to cell dysfunction and damage in hepatocytes8. Increased intracellular Ca2+ activates calpains, which are Ca2+-dependent proteases9 that have been shown to regulate anti-apoptotic proteins including Bcl-210. Previous studies have shown that LPS-induced apoptosis is dependent on changes in Bcl-2 levels11,12. Based on these results, we hypothesized that LPS-induced atherosclerosis is mediated by calpain-induced endothelial cell apoptosis. Our results provide new explanations of the underlying mechanisms of LPS-induced endothelial cell apoptosis and the anti-atherosclerosis effect of amiloride.
Materials and methods
Materials
Amiloride and ZLLal (benzyloxycarbonyl-leucyl-leucinal) were purchased from Biomol Research Laboratories, PA, USA. The calcium chelator, BAPTA, was purchased from Invitrogen Corporation. 2-Carboxyethyl-5(6)-carboxyfluorescein (BCECF) was purchased from Calbiochem (USA). Other chemicals were purchased from Sigma Chemical Co, USA. Anti-Bak, Bcl-2, Bax, β-actin, and secondary antibodies were purchased from Cell Signaling Technology, Inc(Danvers, MA, USA).
Mice
Male apolipoprotein E knockout (ApoE−/−) mice were purchased from Jackson Laboratories (Bar Harbor, ME, USA). 6- to 8-week-old mice were used in the experiments. All of the mice were fed high-fat diets with or without 0.5% (w/w) amiloride for 4 weeks and LPS (10 ug/mouse) infusion into caudal veins. The investigation conformed to the Guide for the Care and Use of Laboratory Animals published by Jilin University.
Cell culture
HUVECs were purchased from ATCC and cultured according to the method described previously13. HUVECs were grown in EBM (Clonetics Inc, Walkersville, MD, USA) supplemented with 12.5 mg/mL ECGF, 10% FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 1 mg/mL hydrocortisone. Cells were subcultured when 80%–90% confluent. Cells in passages 3–8 were used for all of the experiments.
Western blot
HUVECs were lysed in 1×cell-lysis buffer (Cell Signaling Technology, Inc, Danvers, MA, USA), and the protein content was determined by using a BCA protein assay reagent (Pierce, USA). Approximately 20–40 μg of protein was separated by 8%–10% SDS-PAGE and then transferred to nitrocellulose membrane. The membrane was incubated with a 1:1000 dilution of primary antibody overnight at 4 °C, followed by a 1:2000 dilution of horseradish peroxidase-conjugated secondary antibody for 2 h at room temperature. The protein bands were visualized by enhanced chemiluminescence (Amersham Biosciences). The band densities were measured by densitometry (model GS-700, Imaging Densitometer; Bio-Rad). The background was subtracted from the calculated area.
RT-PCR analysis for Bcl-2 mRNA
Total RNA was isolated by a TRIzol (Life Technologies, USA) reagent according to the manufacturer's protocol14. The primer sequences used are as follows: Bcl-2 sense, 5′-GTGGATGACTGAGTACCTGAACC-3′; Bcl-2 antisense, 5′-AGCCAGGAGAAATCAAACAGAG-3′; GAPDH sense, 5′-TCATTTCCTGGTATGACAACG-3′; and GAPDH antisense, 5′-TTACTCCTTGGAGGCCATGT-3′. The PCR protocol for Bcl-2 is consisted of (1) 94 °C initial denaturation for 5 min, (2) 30 cycles of 30 s at 94 °C, 30 s at 55 °C and 2 min at 72 °C, (3) final extension at 72 °C for 15 min. The protocol for GAPDH was the same as Bcl-2 except for 1 min annealing at 65 °C and amplification for 25 cycles.
Measurement of NHE1 activity in HUVECs
The NHE1 activity in HUVECs or aortic tissue in situ was assayed using a NH4Cl pulse method15. Briefly, cells were washed with HCO3−-free HBS buffer (140 mmol/L NaCl, 1 mmol/L CaCl2, 5 mmol/L KCl, 1 mmol/L MgCl2, 6 mmol/L HEPES, 5 mmol/L glucose, pH 7.4). After incubating the HUVECs with HBS containing 10 μmol/L BCECF at 37 °C for 30 min and removing the free-BCECF by washing with HBS, 40 mmol/L of NH4Cl was added to the HBS, incubated for 5 min, and washed out with Na+-free HBS buffer. At this stage, cells were acid-loaded, and the pHi level was decreased. When 100 mmol/L of NaCl was added, the intracellular H+ was pumped out via NHE1, and the pHi increased linearly. This initial rate of pHi recovery (dpHi/dt) reflects the NHE1 activity.
Determination of calpain activity
We used the fluorogenic peptide Suc-Leu-Leu-Val-Tyr-AMC to assay calpain activity as described previously with some modifications16. Briefly, cells were cultured in 24-well plates and washed with PBS, and 80 μmol/L of fluorogenic peptide was added. The fluorescence intensity was immediately recorded at 2-min intervals for 20 min at an excitation wavelength of 360 nm and emission wavelength of 460 nm with a Synergy HT Multi-Detection Microplate Reader (BIO-TEK Instruments Inc, Winooski, USA). We used the initial rate of peptidyl-AMC hydrolysis as the activity of the calpain enzyme.
Measurement of Ca2+i concentration
The intracellular Ca2+i level was assayed using a Fluo-4 NW kit (Invitrogen), as described previously17. Shortly after treatment, the medium was aspirated, the cells were washed with HEPES buffer (pH 7.4), and 1 mL of HEPES buffer containing fluorescent dye was added to the cultured cells. After 30 min of incubation, the fluorescence intensity was measured with an excitation/emission wavelength ratio of 485/520 nm.
Apoptosis assay by TUNEL in HUVECs
HUVECs were washed with cold PBS and then fixed with 4% paraformaldehyde in PBS overnight. Cell apoptosis was detected by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling staining (TMR red) using a kit (Roche Applied Science) and following the provided instruction manual18. The percentage of apoptosis was calculated from the number of terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling-positive cells divided by the total number of cells counted.
DNA fragmentation assay in HUVECs
DNA from HUVECs was extracted with a Quick Apoptosis DNA Ladder Detection Kit (MBL, Nagoya, Japan). The samples were then loaded onto 1.5% agarose gel. After electrophoresis, the gel was subsequently stained with ethidium bromide and visualized under UV illumination19.
Atherosclerotic lesion analysis
The aortic tissue up to the ileal bifurcation was removed and fixed in 4% paraformaldehyde for 16 h. The adventitia was then thoroughly removed under a dissecting microscope. For each mouse, four consecutive sections were stained with Hematoxylin and Eosin (HE). The plaque images were captured using an Olympus microscope connected to a QImaging Retiga CCD camera. The aortic lesion size in each animal was obtained by averaging the lesion areas in the four sections. The lesion area, from the aortic arch to 5 mm distal to the left subclavian artery, was quantified using the Alpha Ease FC software (Version 4.0, Alpha Innotech, Miami, USA).
Immunohistochemistry
The aortic section was dissected, fixed in 4% paraformaldehyde for 16 h, and embedded in paraffin. Four micron-thick sections were deparaffinized, rehydrated, and microwaved in citrate buffer for antigen retrieval. The sections were successively incubated in endogenous peroxidase and alkaline phosphatase block buffer (DAKO), protein block buffer, and primary antibodies, with the antibody incubation being performed overnight at 4 °C. After rinsing in wash buffer, the sections were incubated with labeled polymer-horseradish peroxidase anti-mouse or anti-rabbit antibodies and DAB chromogen. Alternatively, they were incubated with polymer-alkaline phosphatase anti-mouse or anti-rabbit antibody and Permanent Red chromogen (EnVision™ G|2 Doublestain System, DAKO). After the final washes, the sections were counterstained with hematoxylin.
Statistical analysis
The statistical analyses were performed using the SPSS10.0 software. The data are expressed as the mean±SEM. The statistical significance of differences was evaluated by using one-way ANOVA followed by Student's t-test. P<0.05 was considered statistically significant.
Results
LPS induces increased activation of NHE1 activity and intracellular calcium
Earlier studies have shown that LPS induces endothelial cell apoptosis and endothelial dysfunction, which contributes to cardiovascular disease20. However, the mechanisms leading to LPS-induced endothelial cell apoptosis are not well-defined. We first determined whether LPS could activate NHE1 in HUVECs. As shown in Figure 1A, LPS stimulation of HUVECs induced an increase in NHE1 activity beginning at 4 h and peaking at 16 h post-stimulation. NHE1 activity remained elevated at 48 h.

LPS increases amiloride-sensitive NHE1 activity, intracellular calcium, and calpain activity. (A) HUVECs were incubated with LPS (100 ng/mL) from 1 to 48 h. NHE1 activity was assayed by an NH4Cl pulse method plus BCECF dye at the indicated time points. (B) HUVECs were incubated with LPS (100 ng/mL) in the presence or absence of amiloride (1, 5, and 10 μmol/L) for 24 h and the NHE1 activity was measured. (C) HUVECs were incubated with LPS (100 ng/mL) from 1 to 24 h and the intracellular calcium concentration determined by Fluo-4 fluorescence. (D) HUVECs were incubated with or without LPS (100 ng/mL) in the presence or absence of amiloride (10 μmol/L) and the intracellular calcium concentration was determined. (E) HUVECs were incubated with or without LPS (100 ng/mL) for 24 h in the absence or presence of amiloride (10 μmol/L) and the calpain activity was assayed with a fluorogenic peptide substrate. (F) HUVECs were incubated with or without LPS (100 ng/mL) for 24 h in the absence or presence of BAPTA (0.5 mmol/L) and the calpain activity was measured. Data are expressed as the mean±SEM (n=5). bP<0.05 vs control. eP<0.05 vs LPS alone.
The activation of NHE1 increases Na+i, which leads to intracellular Ca2+ overload via the Na+/Ca2+ exchanger and plays a crucial role in many diseases, including ischemia21 and atherosclerosis22. We further investigated whether the activation of NHE1 by LPS resulted in increased intracellular calcium levels. As shown in Figure 1C, LPS increased intracellular calcium levels in HUVECs in a time-dependent manner. Thus, LPS induces increases in NHE1 activity that is associated with significant increases in intracellular calcium in HUVECs.
Amiloride prevents the induction of NHE1 and increases in intracellular calcium
To confirm the specific activation of NHE1 by LPS, we treated HUVECs with amiloride, a well-known NHE1 inhibitor. As shown in Figure 1B, amiloride prevented LPS-induced increases in NHE1 activity in a dose-dependent manner. Additionally, the LPS-induced increase in intracellular calcium was reversed by amiloride treatment (Figure 1D). These data confirm that the LPS-induced increase in intracellular calcium depends on NHE1.
LPS-enhanced calpain activity is both NHE1 and calcium dependent
Calpains are a family of Ca2+-dependent cysteine proteases found in mammals and many lower organisms17,23. In the presence of elevated Ca2+i concentration, calpains are activated and regulate cellular function by degrading many intracellular proteins24,25. Given the LPS-induced increases in intracellular calcium, we determined whether LPS also induced calpain activity in HUVECs. As seen in Figure 1E and 1F, LPS stimulation of HUVECs induced significant increases in calpain activity. Inhibition of NHE1 by amiloride or chelation of intracellular Ca2+ by BAPTA effectively abolished LPS-induced increases in calpain activity. Taken together, these data indicate that LPS induces activation of NHE1, resulting in the elevation of Ca2+i and causing the activation of calpains.
Inhibition of NHE1 by amiloride abolishes LPS-induced down-regulation of Bcl-2 protein
The anti-apoptotic members of the Bcl family (Bcl-xL, Bcl-2, etc) play important roles in regulating cell death26,27. We determined whether LPS-increased calpain activity induced the degradation of Bcl-2. As shown in Figure 2A, HUVECs treated with LPS significantly reduced Bcl-2 levels compared to control cells. The inhibition of NHE1 by amiloride treatment abolished the LPS-induced decrease in Bcl-2 protein levels. Amiloride treatment with LPS prevented the reduction in Bcl-2 protein, suggesting that NHE1 activity is required to decrease Bcl-2 levels.

LPS induces NHE1 and calpain-dependent degradation of Bcl-2. (A) and (B) HUVECs were incubated with or without LPS (100 ng/mL) for 24 h in the absence or presence of amiloride (10 μmol/L). Cells were used to detect (A) Bcl-2 protein levels by western blot and (B) Bcl-2 mRNA levels by RT-PCR. (C) and (D) HUVECs were incubated with or without LPS (100 ng/mL) for 24 h in the absence or presence of the calpain inhibitor ZLLal (50 μmol/L). Cells were used to detect (C) Bcl-2 and (D) Bax and Bak protein levels by Western blot. Mean±SEM. n=3. bP<0.05 vs control. eP<0.05 vs LPS alone.
The reductions in Bcl-2 protein levels could be due to a LPS-induced repression of Bcl-2 gene transcription. To exclude this possibility, we determined the mRNA levels of Bcl-2 during HUVEC treatment with LPS. As indicated in Figure 2B, LPS stimulation failed to alter Bcl-2 mRNA levels. Amiloride had no effect on the Bcl-2 mRNA level. These data suggest that LPS decreases Bcl-2 protein levels, most probably via protein degradation rather than by decreasing the transcription activity of Bcl-2.
LPS-induced reduction in Bcl-2 is inhibited by the calpain inhibitor, ZLLal
To determine whether LPS-induced Bcl-2 degradation is mediated by calpain, we treated HUVECs with LPS in the absence or presence of the calpain inhibitor, ZLLal. As shown in Figure 2C, the inhibition of calpain by ZLLal prevented LPS-induced decreases in Bcl-2 protein levels. These data indicate that LPS-induced reductions in Bcl-2 are calpain-dependent.
It is reported that other apoptosis-related genes, such as the pro-apoptotic genes Bax and Bak, are involved in cell apoptosis28. To investigate whether both Bax and Bak also play roles in LPS-induced apoptosis of HUVECs, we determined the levels of Bax and Bak in the presence or absence of LPS. As shown in Figure 2D, Bax and Bak levels remained stable in the presence or LPS or ZLLal. Thus, Bcl-2 is uniquely degraded in a calpain-dependent manner in LPS-treated HUVECs.
LPS-induced endothelial cell apoptosis requires NHE1, calcium mobilization, and calpain activation
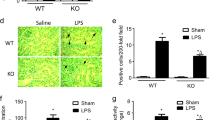
The apoptosis of endothelial cells is very important in the process of atherosclerosis. Because LPS induced the degradation of the anti-apoptotic protein Bcl-2, we determined the apoptosis rates in LPS-treated HUVECs. As shown in Figure 3A and 3B, LPS induced significant increases in HUVEC apoptosis compared with PBS-treated controls, as indicated by positive TUNEL staining. The LPS-induced apoptosis was further confirmed by DNA fragmentation (Figure 3D). The inhibition of calpain activation by amiloride, BAPTA, or ZLLal significantly inhibited LPS-induced apoptosis of HUVECs while having no effect on control cells (Figures 3A and 3B). Amiloride, BAPTA, and ZLLal also effectively prevented LPS-induced DNA fragmentation (Figure 3C). These data indicate that LPS-induced HUVEC apoptosis depends on NHE1 activation, increases in intracellular calcium, and calpain activation.

LPS-induced HUVECs apoptosis depends on NHE1, calcium, and calpain. HUVECs were incubated with or without LPS (100 ng/mL) for 24 h in the absence or presence of amiloride (10 μmol/L), BAPTA (0.5 mmol/L), or ZLLal (50 μmol/L). (A) HUVEC cell apoptosis was assayed by TUNEL staining. Scale bar=20 μm. (B) Quantification of TUNEL staining. (C) Detection of DNA fragments. Mean±SEM. n=5. bP<0.05 vs control. eP<0.05 vs LPS alone.
Amiloride attenuates LPS-accelerated atherosclerosis in ApoE−/− mice
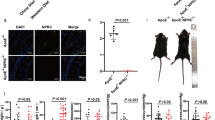
Our data suggest that amiloride may have significant effects on LPS-induced atherosclerosis by inhibiting NHE1 activation and endothelial apoptosis. To test this hypothesis, we investigated whether amiloride could prevent LPS-induced atherosclerosis in vivo. ApoE−/− mice on a high-fat diet were treated with or without LPS and with or without 0.5% amiloride (w/w) for 4 weeks. Compared with control ApoE−/− mice on a high-fat diet, LPS remarkably accelerated atherosclerotic lesion formation in the aorta (Figure 4A). Amiloride suppressed the accelerated atherosclerotic lesions in LPS-treated mice. To assess endothelial apoptosis, we performed immunohistochemistry for Bcl-2 in representative aortic lesions. As shown in Figure 4B, LPS decreased the expression of Bcl-2, whereas amiloride treatment in the presence of LPS led to significant increases in the expression of Bcl-2.

Amiloride administration suppresses LPS-accelerated atherosclerosis in ApoE−/− mice. ApoE−/− mice were put on a high-fat diet for 6 weeks then treated with or without LPS. Additional cohorts were treated with or without 0.5% amiloride (w/w). (A) HE staining of aortic tissue. Scale bar=100 μm. (B) Bcl-2 protein expression by immunohistochemistry. Scale bar=20 μm. (C) NHE1 activity in mouse aorta extracts. (D) Proposed scheme of LPS-induced endothelial injury and atherosclerosis with amiloride prevention. Mean±SEM. n=5. bP<0.05 vs control. eP<0.05 vs LPS alone.
To confirm whether LPS activates NHE1 in vivo, we determined the activity of NHE1 in extracts of mouse aorta tissue. As shown in Figure 4C, LPS significantly increased the activity of NHE1 within the aorta. The administration of amiloride attenuated this LPS-induced increase in NHE1 activity. Surprisingly, NHE1 activity in control mice was unchanged with amiloride treatment. Taken together, our data indicate that LPS-accelerated atherosclerosis may occur via activation of NHE1 leading to changes in Ca2+i, activation of calpain, and degradation of Bcl-2, thus driving endothelial apoptosis and lesion formation. Inhibiting the NHE1 pathway by amiloride effectively suppressed these downstream effects and prevented LPS-induced atherosclerosis.
Discussion
The current study demonstrates that amiloride attenuates LPS-accelerated atherosclerosis by inhibiting NHE1-dependent calpain-mediated Bcl-2 degradation in endothelial cells. The inhibition of NHE1 by amiloride reversed LPS-induced increases in intracellular calcium and calpain activity. In addition, the inhibition of either NHE1 by amiloride or calpain by ZLLal blocked Bcl-2 degradation caused by LPS. These results strongly suggest that NHE1 is required for LPS-induced endothelial cell apoptosis and the anti-atherosclerosis effects of amiloride.
Endothelial cell injury is common in the pathogenesis of several diseases, including atherosclerosis29, hypertension30, and congestive heart failure31. During bacterial sepsis, endothelial cell death is the final step of a process that begins when quiescent endothelial cells are induced to express pro-coagulant, pro-adhesive, and vasoconstrictive factors. Results from this study demonstrate that LPS induces apoptosis in HUVECs in a NHE1- and calpain-dependent manner. These findings agree with those obtained in other species, both in vitro and in vivo32,33.
Our data clearly indicate that LPS-induced apoptosis is mediated by NHE1 via the calcium/calpain pathway. Calpains are a family of calcium-dependent proteases that act independently of the proteasome pathway and cleave a number of cellular substrates, including kinases, phosphatases, transcription factors, and cytoskeletal proteins34. In this study, we found that LPS increased the intracellular calcium and calpain activity in vascular endothelial cells. The chelation of intracellular free-calcium by BAPTA effectively prevented calpain activation and induction of apoptosis by LPS.
Our data also indicate that LPS-induced NHE1 activation is effectively inhibited in vitro and in vivo by amiloride. One potential mechanism of the LPS-induced increase of NHE1 activity may involve either a phosphorylation-dependent increase in the activity of existing exchangers or the activation of dormant membrane-associated exchangers. In fact, Sardet et al35, have demonstrated that NHE1 is rapidly phosphorylated in response to various mitogens and concluded that this phosphorylation of NHE1 is temporally correlated with its activation. Additional studies are needed to determine the precise mechanism of NHE1-activation by LPS and how amiloride alters this activation.
The anti-apoptotic Bcl-2 protein is localized to outer mitochondrial membrane and functions to prevent cytochrome c release from mitochondria36. Here, we found that LPS induced reductions in Bcl-2 protein levels in HUVECs that could be prevented by treatment with amiloride or calpain inhibitor ZLLal; the mRNA levels of Bcl-2 were not affected. All of the results indicate that calpain participates in the LPS-induced degradation of Bcl-2; however, it remains unclear which calpain isoform is involved and whether Bcl-2 is the direct substrate of calpain in LPS-treated HUVECs.
In conclusion, the inhibition of NHE1 by amiloride completely blocked LPS-induced atherosclerosis in mice and is mediated by calcium and calpain-dependent Bcl-2 degradation in HUVECs (Figure 4D). The inhibition of NHE1 could be a promising novel approach to halt or even reverse atherosclerosis in patients.
Author contribution
Gui-mei CUI, Yu-xi ZHAO, and Na-na ZHANG performed the research; Zeng-shan LIU and Wan-chun SUN analyzed the data; and Qi-sheng PENG designed the research, analyzed the data, and wrote the paper.
References
Teiwes J, Toto RD . Epithelial sodium channel inhibition in cardiovascular disease. A potential role for amiloride. Am J Hypertens 2007; 20: 109–17.
Raetz CR . Biochemistry of endotoxins. Annu Rev Biochem 1990; 59: 129–70.
Liu T, Huang Y, Likhotvorik RI, Keshvara L, Hoyt DG . Protein Never in Mitosis Gene A Interacting-1 (PIN1) regulates degradation of inducible nitric oxide synthase in endothelial cells. Am J Physiol Cell Physiol 2008; 295: C819–27.
Cybulsky MI, Chan MK, Movat HZ . Acute inflammation and microthrombosis induced by endotoxin, interleukin-1, and tumor necrosis factor and their implication in gram-negative infection. Lab Invest 1988; 58: 365–78.
Pober JS, Cotran RS . The role of endothelial cells in inflammation. Transplantation 1990; 50: 537–44.
Choi KB, Wong F, Harlan JM, Chaudhary PM, Hood L, Karsan A . Lipopolysaccharide mediates endothelial apoptosis by a FADD-dependent pathway. J Biol Chem 1998; 273: 20185–8.
Wang SX, Xiong XM, Song T, Liu LY . Protective effects of cariporide on endothelial dysfunction induced by high glucose. Acta Pharmacol Sin 2005; 26: 329–33.
Wang D, Dou K, Song Z, Liu Z . The Na+/H+ exchange inhibitor: a new therapeutic approach for hepatic ischemia injury in rats. Transplant Proc 2003; 35: 3134–5.
Goll DE, Thompson VF, Li H, Wei W, Cong J . The calpain system. Physiol Rev 2003; 83: 731–801.
Gil-Parrado S, Fernandez-Montalvan A, Assfalg-Machleidt I, Popp O, Bestvater F, Holloschi A, et al. Ionomycin-activated calpain triggers apoptosis. A probable role for Bcl-2 family members. J Biol Chem 2002; 277: 27217–26.
Schelling JR, Abu Jawdeh BG . Regulation of cell survival by Na+/H+exchanger-1. Am J Physiol Renal Physiol 2008; 295: F625–32.
Garciarena CD, Caldiz CI, Portiansky EL, Chiappe de Cingolani GE, Ennis IL . Chronic NHE-1 blockade induces an antiapoptotic effect in the hypertrophied heart. J Appl Physiol 2009; 106: 1325–31.
Wang S, Xu J, Song P, Wu Y, Zhang J, Chul Choi H, et al. Acute inhibition of guanosine triphosphate cyclohydrolase 1 uncouples endothelial nitric oxide synthase and elevates blood pressure. Hypertension 2008; 52: 484–90.
Kyronlahti A, Ramo M, Tamminen M, Unkila-Kallio L, Butzow R, Leminen A, et al. GATA-4 regulates Bcl-2 expression in ovarian granulosa cell tumors. Endocrinology 2008; 149: 5635–42.
Wang SX, Sun XY, Zhang XH, Chen SX, Liu YH, Liu LY . Cariporide inhibits high glucose-mediated adhesion of monocyte-endothelial cell and expression of intercellular adhesion molecule-1. Life Sci 2006; 79: 1399–404.
Dong Y, Tan J, Cui MZ, Zhao G, Mao G, Singh N, et al. Calpain inhibitor MDL28170 modulates Abeta formation by inhibiting the formation of intermediate Abeta46 and protecting Abeta from degradation. Faseb J 2006; 20: 331–3.
Wang S, Peng Q, Zhang J, Liu L . Na+/H+ exchanger is required for hyperglycemia-induced endothelial dysfunction via calcium-dependent calpain. Cardiovasc Res 2008; 80: 255–62.
Song P, Xie Z, Wu Y, Xu J, Dong Y, Zou MH . Protein kinase Czeta-dependent LKB1 serine 428 phosphorylation increases LKB1 nucleus export and apoptosis in endothelial cells. J Biol Chem 2008; 283: 12446–55.
Nakatani K, Takeshita S, Tsujimoto H, Sekine I . Intravenous immunoglobulin (IVIG) preparations induce apoptosis in TNF-alpha-stimulated endothelial cells via a mitochondria-dependent pathway. Clin Exp Immunol 2002; 127: 445–54.
Bannerman DD, Goldblum SE . Mechanisms of bacterial lipopolysaccharide-induced endothelial apoptosis. Am J Physiol Lung Cell Mol Physiol 2003; 284: L899–914.
Toda T, Kadono T, Hoshiai M, Eguchi Y, Nakazawa S, Nakazawa H, et al. Na+/H+ exchanger inhibitor cariporide attenuates the mitochondrial Ca2+ overload and PTP opening. Am J Physiol Heart Circ Physiol 2007; 293: H3517–23.
Koliakos G, Befani C, Paletas K, Kaloyianni M . Effect of endothelin on sodium/hydrogen exchanger activity of human monocytes and atherosclerosis-related functions. Ann N Y Acad Sci 2007; 1095: 274–91.
Azuma M, Shearer TR . The role of calcium-activated protease calpain in experimental retinal pathology. Surv Ophthalmol 2008; 53: 150–63.
Stalker TJ, Gong Y, Scalia R . The calcium-dependent protease calpain causes endothelial dysfunction in type 2 diabetes. Diabetes 2005; 54: 1132–40.
Stalker TJ, Skvarka CB, Scalia R . A novel role for calpains in the endothelial dysfunction of hyperglycemia. Faseb J 2003; 17: 1511–3.
Lessene G, Czabotar PE, Colman PM . BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov 2008; 7: 989–1000.
Yip KW, Reed JC . Bcl-2 family proteins and cancer. Oncogene 2008; 27: 6398–406.
Fletcher JI, Huang DC . Controlling the cell death mediators Bax and Bak: puzzles and conundrums. Cell Cycle 2008; 7: 39–44.
Chen K, Keaney J . Reactive oxygen species-mediated signal transduction in the endothelium. Endothelium 2004; 11: 109–21.
Chen M, Bao W, Aizman R, Huang P, Aspevall O, Gustafsson LE, et al. Activation of extracellular signal-regulated kinase mediates apoptosis induced by uropathogenic Escherichia coli toxins via nitric oxide synthase: protective role of heme oxygenase-1. J Infect Dis 2004; 190: 127–35.
Cines DB, Pollak ES, Buck CA, Loscalzo J, Zimmerman GA, McEver RP, et al. Endothelial cells in physiology and in the pathophysiology of vascular disorders. Blood 1998; 91: 3527–61.
Frey EA, Finlay BB . Lipopolysaccharide induces apoptosis in a bovine endothelial cell line via a soluble CD14 dependent pathway. Microb Pathog 1998; 24: 101–9.
McCuskey RS, Urbaschek R, Urbaschek B . The microcirculation during endotoxemia. Cardiovasc Res 1996; 32: 752–63.
Sorimachi H, Ishiura S, Suzuki K . Structure and physiological function of calpains. Biochem J 1997; 328: 721–32.
Sardet C, Counillon L, Franchi A, Pouyssegur J . Growth factors induce phosphorylation of the Na+/H+ antiporter, glycoprotein of 110 kD. Science 1990; 247: 723–6.
Green DR, Reed JC . Mitochondria and apoptosis. Science 1998; 281: 1309–12.
Acknowledgements
We thank Dr Humphrey (Department of Medicine, University of Oklahoma Health Sciences Center) for critically reading this manuscript. This work was funded by Jilin University grant (450060445662, 430504001043, and 430505010272 to Qi-sheng PENG; 4305050102Q1 to Wan-chun SUN).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Cui, Gm., Zhao, Yx., Zhang, Nn. et al. Amiloride attenuates lipopolysaccharide-accelerated atherosclerosis via inhibition of NHE1-dependent endothelial cell apoptosis. Acta Pharmacol Sin 34, 231–238 (2013). https://doi.org/10.1038/aps.2012.155
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2012.155
Keywords
This article is cited by
-
SGLT2 inhibitors prevent LPS-induced M1 macrophage polarization and alleviate inflammatory bowel disease by downregulating NHE1 expression
Inflammation Research (2023)
-
Novel Anti-inflammatory Effects of Canagliflozin Involving Hexokinase II in Lipopolysaccharide-Stimulated Human Coronary Artery Endothelial Cells
Cardiovascular Drugs and Therapy (2021)
-
Na+-H+ exchanger 1 determines atherosclerotic lesion acidification and promotes atherogenesis
Nature Communications (2019)
