
Abstract
Aim:
To investigate the safety, tolerability and pharmacokinetics of intravenous hemoporfin, a novel photosensitive drug for the treatment of port-wine stain (PWS), in healthy Chinese volunteers following single-dose administration.
Methods:
Thirty-six healthy Chinese subjects were enrolled. The subjects were administered hemoporfin (2.5, 5, 7.5 or 10 mg/kg) via single-dose intravenous infusion. Pharmacokinetics of the drug were studied in the groups with doses of 2.5, 5 and 7.5 mg/kg, and tolerability was studied in all the 4 groups. Safety and tolerance were evaluated by monitoring adverse events and laboratory parameters, and pharmacokinetics were assessed by determining hemoporfin content with a validated high-performance liquid chromatography with fluorescence detection (HPLC/FLD) method.
Results:
Mild and transient adverse events occurred in the trial (n=10), but none were serious, and no subjects were withdrawn from the trial. The gastrointestinal tract adverse events, such as nausea, stomach upset, abdominal pain and vomiting, were observed in the groups with doses of 7.5 and 10 mg/kg. Increased alanine aminotransferase (ALT) concentration was found in 3 subjects, and increased alkaline phosphatase (ALP) concentration in one subject. The half-life of hemoporfin for doses of 2.5, 5, and 7.5 mg/kg was 1.26 h, 1.31 h, and 1.70 h, respectively. Cmax and AUC increased with dose for intravenous single-dose administration of hemoporfin in the 2.5, 5, and 7.5 mg/kg groups. Urinary excretion of hemoporfin within 12 h was less than 0.2%.
Conclusion:
Hemoporfin is safe and well-tolerated in healthy Chinese volunteers at a single intravenous dose of up to 10 mg/kg. It was rapidly cleared from the blood and had a short half-life, which insures a short light-avoidance period.
Similar content being viewed by others


Introduction
Port-wine stain (PWS) is a congenital vascular malformation of the dermis that affects 0.3% to 0.5% of newborns and is commonly associated with psychosocial impairment1. In China, photodynamic therapy (PDT) has been applied for the treatment of PWS since 19912. In this therapy, an exogenous chromophore-like porphyrin derivative is administered in the blood and concentrates in the ectatic capillaries. Subsequent irradiation using light of appropriate wavelength generates reactive oxygen species (ROS) in the presence of porphyrin derivatives and oxygen, selectively damaging the capillary wall3. PDT has been shown to be effective for PWS4, 5, 6. However, previously used photosensitive drugs, such as hematoporphyrin derivative (HpD) or Photocarcinorin, are complex mixtures of porphyrins containing more than 10 chemicals, with a long elimination half-life of >30 h7. This led to prolonged systemic photosensitivity to visible light, which lasted for one to three months following drug administration, limiting the clinical application of PDT2, 8.
Hemoporfin (hematoporphyrin monomethyl ether) is a novel porphyrin-related photosensitive drug first developed in China. Hemoporfin consists of two positional isomers of 7(12)-(1-methoxyethyl)-12(7)-(1-hydroxyethyl)-3,8,13,17-tetramethyl-21H,23H-porphin-2,18-di-propionic acid (Figure 1). Previous reports have shown that hemoporfin has strong photodynamic effects and hemoporfin-based PDT induces significant cell death9, 10. An experimental study using chicken comb as an animal model of PWS showed that hemoporfin-based PDT could selectively destroy the microvasculature of the superficial portion of the dermis and thus cause the red combs to completely fade without damaging the overlying skin11. Pharmacokinetic studies utilizing hemoporfin have been performed in various animals. Studies in rats showed that the drug was distributed rapidly to tissues, particularly to the liver, stomach, intestines, and spleen (JG Sun and GJ Wang, unpublished data). Hemoporfin was rapidly transferred from the central compartment to the peripheral compartment in rabbits following intravenous injection. Data for the elimination phase showed that hemoporfin was quickly eliminated (the elimination half-life was approximately 2.5 h) and not accumulated12. These preclinical data indicate that hemoporfin could achieve satisfactory clinical results and enable the use of shorter light-avoidance time periods during the course of therapy.

Chemical structure of hemoporfin.
This Phase I clinical trial of hemoporfin was approved by the State Food and Drug Administration of China (SFDA, 2005L01082). The objectives of the present study were to assess the safety, tolerance and pharmacokinetics of hemoporfin in healthy Chinese volunteers. Results of this trial are herein reported to provide evidence for the next phase of clinical trials.
Materials and methods
This study was performed at Base for Drug Clinical trial and approved by the ethics committee of the First Hospital of Peking University, and conducted in accordance with the Declaration of Helsinki and Good Clinical Practice.
Subjects
Subjects (of Han nationality) were enrolled according to the following criteria: (i) 18–45 years old with body mass index (BMI) of 19–27 kg/m2; (ii) normal vital signs, physical and laboratory findings, negative for HIV and hepatitis B/C, no abnormalities on ECG, no history of heart, liver, renal, digestive system disorders, nervous or psychological disease or metabolic disorders; (iii) no history of allergy to any components of hemoporfin for injection; (iv) no smoking, drinking or drug addiction prior to or during the study; (v) no pregnancy or lactation for female subjects; (vi) no drugs or other medication, or participation in any clinical trial that might interfere with absorption, distribution, metabolism or excretion of hemoporfin or be harmful to human organs three months prior to or during the study; and (vii) voluntary signature on an informed consent form. Subjects with systemic disease or inherited/acquired coagulation defects were excluded.
Drug
Hemoporfin for injection was formulated and supplied by Shanghai Fudan-Zhangjiang Bio-Pharmaceutical Co, Ltd (Shanghai, China). It was a sterile lyophilized powder containing 100 mg of hemoporfin per vial.
Study design
A randomized, open-label, dose-escalation study was conducted to assess the safety and tolerance of hemoporfin. Thirty-six healthy Chinese subjects were enrolled, ten subjects each in the 2.5, 5, and 7.5 mg/kg dose groups and six subjects in the 10 mg/kg dose group. The trial was designed to begin with the 2.5 mg/kg dose group and not proceed to the higher dose group until the safety and tolerance to the lower doses were confirmed. Within each cohort, toxicity was considered unacceptable if any subject experienced a severe adverse event or half of the subjects in any dose group experienced mild adverse events that were attributed to the study medication and were not resolved within a reasonable time period. Pharmacokinetics of hemoporfin were studied in the 2.5, 5, and 7.5 mg/kg dose groups, and tolerability was studied in all four groups. All subjects were required to fast overnight (10 h) before and for 4 h following administration. After drug administration, patients were instructed to avoid exposure to sunlight or bright indoor light for one week.
The medication was prepared in normal saline (60 mL) and was administered by iv infusion over a 20-min period, using an injection pump at a constant speed. The solution was protected from light and used within 4 h after preparation. A volume of 2 to 4 mL of normal saline was intravenously injected following the end of infusion of the drug to reduce drug concentration in the vessels. This prevented the occurrence of photosensitive reactions at the injection site.
Safety and tolerance
All subjects were maintained in the study unit and continuously observed. The variables used to assess safety and tolerability included adverse events, physical examination, vital signs, and 12-lead electrocardiogram, routine blood and urine test, coagulation test and blood biochemical test. The blood biochemical test included urea nitrogen (BUN), creatinine (Cr), uric acid (UA), total protein (TP), albumin (ALB), albumin/globulin ratio (A/G), alanine aminotransferase (ALT), aspartic transaminase (AST), γ-glutamyltransferase (GGT), alkaline phosphatase (ALP), total bilirubin (TBIL), direct bilirubin (DBIL), fasting blood glucose (GLU), triglyceride (TG) and total cholesterol (TCHO). Safety tests were conducted 24 to 48 h before and on days 1, 2, 3, and 7 after drug administration.
Adverse event data were obtained voluntarily from subjects and by daily monitoring and questioning of subjects by study personnel. The expected adverse event associated with hemoporfin was photosensitivity reaction. All subjects who received hemoporfin are photosensitive and must observe precautions to avoid exposure of skin and eyes to direct sunlight or bright indoor light (eg, from examination lamps, including dental lamps, operating room lamps, unshaded light bulbs at close proximity) for at least seven days. Adverse events were evaluated for their intensity, seriousness, and relationship to the study medication. Adverse events were defined as mild (awareness of a sign or symptom but easily tolerated), moderate (discomfort sufficient to cause interference with normal activities), or severe (incapacitating, with an inability to perform normal activities). Causality between the study drug and an adverse event, described as “certainly,” “probably,” “possibly,” “suspected,” or “not related,” was defined using the criteria developed by the World Health Organization13.
Sample collection
Blood samples (3 mL) were collected into tubes containing ammonium heparinate and centrifuged immediately to obtain plasma samples. These samples were then transferred to polypropylene vials and kept frozen below -20 °C until analyses were conducted. Samples were collected as follows: prior to dosing (0 h); 5, 10, and 20 min during dosing; and 5, 10, 20, 30 min and 1, 1.5, 2, 3, 4, 6, 8 h post-dosing. Urine was collected prior to dosing (10 mL), and was also pooled at 0 to 2, 2 to 4, 4 to 8, and 8 to 12 h post-dosing. Preliminary analyses showed that no analyte was present in urine 12 h following drug administrations. After the total volume of urine for each time range was measured, 10 mL was maintained at -20 °C until analyses were conducted.
Drug analysis
Concentrations of hemoporfin in plasma and urine were determined using high-performance liquid chromatography with fluorescence detection (HPLC/FLD). Fluorescein was used as the internal standard. This method was previously developed and fully validated for determination of hemoporfin in plasma14. In brief, plasma (0.5 mL) and urine samples (diluted) were pretreated using liquid-liquid extraction with ethyl acetate, and the supernatant was evaporated to dryness and reconstituted with 200 μL of mobile phase. The analyte was separated on a Diamonsil C18 column (4.6 mm×150 mm, 5 μm) by isocratic elution with tetrahydrofuran-20 μmol/L sodium acetate buffer (adjusted to pH 6.0 with acetic acid; 40:60, v/v) at a flow rate of 1.0 mL/min and analyzed using a fluorescence detector with the excitation and emission wavelengths set at 395 and 613 nm, respectively. Analyses of blank serum and urine indicated no interference of endogenous components with detection of hemoporfin in the final extract. The weighted (1/x) calibration curve was linear over the plasma concentration range of 0.025–5 μg/mL and urine concentration range of 0.025–1 μg/mL with a correlation coefficient (r) of 0.9982 and 0.9998, respectively. The lower limit of quantification for hemoporfin was 0.025 μg/mL. The inter- and intra-day accuracies and precisions were all within 10%. In plasma, the mean method recoveries were 95.1%–97.3% and the mean extraction recoveries were 55.9%–61.4%. In urine, the mean method recoveries were 92.7%–101.7% and the mean extraction recoveries were 75.9%–79.0%.
Pharmacokinetic calculations
The mean hemoporfin concentration in plasma at each time point and in urine over each interval was determined by averaging of the data, and pharmacokinetic parameters were calculated using Winnonlin 5.0 (Pharsight Corporation; Sunnyvale, CA, USA). Single-dose pharmacokinetic parameters were calculated from plasma concentration-time data by the non-compartment method. Peak plasma concentration (Cmax) and the time to reach peak concentration (Tmax) were recorded from experimental observations. The parameters of the model were used to calculate values for clearance (CL), volume of distribution [V(L)], elimination half-life (t1/2), area under the curve (AUC) from time zero to infinity (AUC0–∞), AUC from time zero to the time of the last quantifiable concentration (AUC0–tn), mean residence time (MRT) from time zero to infinity (MRT0–∞), MRT from time zero to the time of the last quantifiable concentration (MRT0–tn), and the elimination rate constant (Ke). The fraction of the drug excreted in urine (Ae%) was calculated by dividing Ae by the dose administered, where Ae is the cumulative amount of unchanged drug excreted into the urine.
Statistical analyses
The analyses of safety were performed with SPSS (version 11.5). Descriptive statistics were used to summarize demographic data and assess safety variables including adverse events, laboratory assays, vital signs and electrocardiograms. The variations of laboratory parameters before and after drug administration were analyzed using paired t-test and the variances among groups were analyzed using ANOVA. Adverse events are summarized by reported frequencies and percentages.
Results
Study population
Thirty-six (19 male, 17 female) subjects were enrolled in this study. All subjects completed the trials as planned. Demographic characteristics of the study population are presented in Table 1. There were no significant differences in age, weight, height or BMI among the four groups.
Safety and tolerability
A single dose of hemoporfin was well tolerated in these healthy volunteers at doses up to 10 mg/kg, with no severe adverse events occurring during the study, and all subjects were in good compliance. The most commonly reported adverse events were related to the gastrointestinal tract. Nausea (3 subjects, 8.3%), upset stomach (2 subjects, 5.6%), abdominal pain (2 subjects, 5.6%) and vomiting (1 subjects, 2.8%) were observed in the 7.5 mg/kg and 10 mg/kg dose groups. Increased ALT concentrations were found in three subjects, one in the 2.5 mg/kg group and two in the 7.5 mg/kg group. Increased ALP concentration was also found in one subject in the 5 mg/kg group. High ALT and ALP concentrations (ALT<3×ULN, ALP<2×ULN; where ULN=upper limit of normal) were observed on d 4 or d 7 following drug administration but returned to normal on d 14 without additional treatment. The aforementioned adverse events were considered to be related to the study drug; ten of the thirty-six (27.8%) hemoporfin-treated subjects had drug-related adverse events (details shown in Table 2). All adverse events and laboratory abnormalities were mild and tolerable, did not lead to discontinuation of the study, and recovery was achieved without treatment. Blood pressure, pulse rate, body temperature, electrocardiographic findings, physical examination findings, hematology results, and urinalysis results following dosing did not show any clinically relevant differences from baseline.
Pharmacokinetics
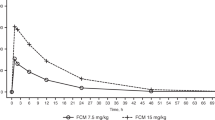
The mean plasma concentration-versus-time profiles after the administration of a single dose of hemoporfin are shown in Figure 2. As expected, Cmax was observed at 20 min following intravenous administration (at the end of the infusion). Plasma hemoporfin levels rapidly decreased, exhibiting a bi-exponential decline with a rapid distribution phase followed by a moderately rapid elimination phase. Hemoporfin was not detectable in most plasma samples after 6 h.

Plasma concentration-time curves of hemoporfin following iv administration of doses of 2.5, 5, 7.5 mg/kg. The data are the means±standard deviations for 10 subjects in each group.
The mean values for the pharmacokinetic parameters of hemoporfin are summarized in Table 3. Hemoporfin had a short half-life (t1/2): 1.26 h, 1.31 h, and 1.70 h for doses of 2.5, 5, and 7.5 mg/kg, respectively, and a short mean residence time (MRT): 12.2 min, 17.7 min, and 26.6 min for doses of 2.5, 5, and 7.5 mg/kg, respectively. For doses of 2.5, 5, and 7.5 mg/kg, mean Cmax values were 17.49, 35.72, and 92.18 mg/L, respectively, and the mean AUC0–∞ values were 6.41, 17.61, and 64.39 mg·h/L, respectively. Cmax and AUC both increased with dosage.
For the 2.5, 5, and 7.5 mg/kg dose groups, urinary excretion of the prototype drug within a 12-h period after drug administration was 0.14%±0.07%, 0.15%±0.03%, and 0.12%±0.05%, respectively.
Significant differences were observed in pharmacokinetic parameters between male and female subjects in the 7.5 mg/kg dose group. Plasma concentrations of hemoporfin in female subjects were higher than those in the male subjects at the same time point following drug administration. For male and female subjects, the mean Cmax values were 67.64±7.97 mg/L and 116.72±20.27 mg/L, respectively; the mean AUC0–tn values were 39.99±4.96 mg·h/L and 88.58±36.00 mg·h/L, respectively; the mean volume values were 30.9±4.9 L and 12.4±4.5 L, respectively; and the mean CL values were 11.5±1.1 L/h and 5.5±1.3 L/h, respectively. There were no significant differences in pharmacokinetic parameters in plasma between male and female subjects in the 2.5 and 5 mg/kg groups.
Discussion
This study showed that iv administration of hemoporfin at doses from 2.5 to 10 mg/kg is safe and well tolerated. Adverse events were generally mild in intensity and required no treatment or intervention. Nausea, upset stomach, abdominal pain and vomiting were the most frequently reported adverse events considered to be related to hemoporfin. These events were dose dependent and found only in the high-dose groups of 7.5 mg/kg and 10 mg/kg. Increases in ALT and ALP concentrations were the most frequently reported laboratory abnormalities. They might be caused by individual variances as they were not relevant to the dose and no significant differences among the different groups were found when analyzed using ANOVA.
Hemoporfin is primarily cleared by the liver, with less than 0.2% of the drug excreted in urine. Hemoporfin has a molecular weight of 612.72 and is poorly water soluble, which favor hepatobiliary elimination. A previous pharmacokinetic study in rats showed that following a single intravenous dose of 10 mg/kg hemoporfin, biliary excretion within 12 h was about 81.8%±9.0%. Urinary excretion within 48 h was about 0.19%±0.17%. Fecal excretion within 48 h was about 57.55%± 15.5% (JG Sun and GJ Wang, unpublished data). The results showed that hemoporfin was excreted predominantly into the bile and eventually into the feces.
After iv administration, hemoporfin was distributed rapidly throughout the body during the first 1 to 3 h, followed by an elimination phase with a t1/2 of approximately 1 to 2 h. For the 2.5 and 5 mg/kg dose groups, the mean Cmax and AUC increased proportionately with dosage. For the 7.5 mg/kg dose group, drug metabolism in the plasma of female subjects was different than the male subjects. The mean Cmax and AUC of male subjects increased proportionately with dosage, but the Cmax and AUC values in the female subjects increased significantly to almost twice the values in the male subjects. The V and CL values of the females were half of the males. No significant differences in drug metabolism in plasma were observed between male and female subjects in the 2.5 and 5 mg/kg groups. These data suggested that females given the same high dose would have substantially decreased hemoporfin clearance and higher levels of systemic exposure. The gender differences indicate that female subjects should be observed for variations in drug metabolism and drug reaction when receiving a large dose of hemoporfin.
The presence of the decreased hemoporfin clearance in female subjects at the high dose needs to be confirmed in clinical trials with larger sample sizes. The mechanisms responsible for the phenomenon are still under investigation. Drug transporters such as P-glycoprotein are likely to be involved in the gender differences. P-glycoprotein is an energy-dependent efflux pump responsible for transporting drugs (usually high molecular weight substances) out of cells and can be expressed in normal tissues such as the luminal surface of biliary hepatocytes15. This pump might play an important role in the elimination of hemoporfin to the bile. P-glycoprotein level in female liver is only 30% to 50% of that in male liver16, and P-glycoprotein may be saturated at higher drug concentrations, so the low P-glycoprotein level in female liver may account for the decreased clearance of hemoporfin in females given large doses.
Compared to the 7.5 and 10 mg/kg dose groups, fewer adverse events were found in the two lower-dose groups, as expected. In addition, hemoporfin exhibited linear pharmacokinetics and no gender-dependent differences in the 2.5 and 5 mg/kg dose groups. Based on the above results, we decided to choose the dosage range from 2.5 to 5 mg/kg for future phase II clinical trials.
A goal of PDT for the treatment of PWS is to selectively destroy the abnormal vasculature but spare adjacent normal skin structures. It is very important to determine the light dose and the time between drug dosing and light application. While shining the light at the PWS lesions during or shortly following iv injection of a photosensitizing drug, when the drug is still highly concentrated inside the circulation (including the PWS vessels) and undergoes little diffusion to the surrounding tissue, the photochemically-induced reactive oxygen species (ROS) would most likely be located within the vessels and, therefore, selectively damage the vascular endothelial cells, induce thrombosis and obstruct capillary vessels. Because the amount of photosensitizing drug outside the vessels is low, the vascular-acting, PDT-induced skin lesion is negligible or reversible8. For the 2.5 and 5 mg/kg dose groups, the pharmacokinetics of hemoporfin indicate that within 50 min after infusion, hemoporfin concentration in vessels were maintained at relatively high levels. Thereafter, the vascular concentrations decreased to a low level and it was presumed that the drug achieved a significant extravascular distribution. This suggests that light application should be within certain time limits (ie, 50 min after infusion) in hemoporfin-based PDT for the treatment of PWS.
The short t1/2 of hemoporfin (1–2 h) suggests that no accumulation would be expected with repeated doses at intervals of one week or shorter. The current recommended dosing schedule is every two months for patients with PWS. The rapid elimination of hemoporfin contributed to a short light-avoidance period. In this study, precautions to avoid photosensitivity were required for one week after treatment, and no photosensitivity reaction was observed throughout the study.
In conclusion, hemoporfin is safe and well tolerated in healthy Chinese subjects at a single dose of between 2.5 mg/kg and 10 mg/kg. No serious adverse events occurred during the study and all subjects remained in good compliance. Cmax and AUC values increased with dose for intravenous, single-dose administration of hemoporfin in the 2.5, 5, and 7.5 mg/kg groups. Hemoporfin is rapidly cleared from the blood and has a short t1/2 of <2 h, which supports a short light-avoidance period of one week.
Author contribution
Yi-min CUI, Pei-hong SUN, and Ji-ning TAO designed research; Pei-hong SUN, Xia ZHAO, Ying ZHOU, Yan LIANG, and Hui-lin ZHANG performed research; Xia ZHAO and Ying ZHOU analyzed data; Pei-hong SUN wrote the paper; Yi-min CUI and Ji-ning TAO critically revised the manuscript.
References
Jacobs AH, Walton RG . The incidence of birthmarks in the neonate. Pediatrics 1976; 58: 218–22.
Gu Y, Li J, Shan H, Wang K, Jiang Y, Zhang Y, et al. Clinical application of copper vapor laser in pdt for fifty cases of PWS. Chin J Laser Med Surg 1994; 3: 215–7.
Jasim ZF, Handley JM . Treatment of pulsed dye laser-resistant port wine stain birthmarks. J Am Acad Dermatol 2007; 57: 677–82.
Jiang L, Gu Y, Li X, Zhao X, Li J, Wang K, et al. Changes of skin perfusion after photodynamic therapy for port wine stain. Chin Med J (Engl) 1998; 111: 136–8.
Ouyang T, Cheng Y, Xing X . Clinical application of photodynamic therapy combined with non-coherent light (red light) for treatment of port-wine stains. Chin J Plast Surg Burn 1998; 14: 163–5.
Chen Y, Li H, Liu S . Treatment of nevas flammeus with copper vapor laser in photodynamic therapy. Acta Acad Med Hubei 1999; 20: 162–3.
Xu DY, Editor. Tumor photodynamic therapy. Beijing: Chinese Medical Science and Technology Press; 1996.
Qin ZP, Li KL, Ren L, Liu XJ . Photodynamic therapy of port wine stains-a report of 238 cases. Photodiagn Photodyn Ther 2007; 4: 53–9.
Song K, Kong B, Qu X, Li L, Yang Q . Phototoxicity of hemoporfin to ovarian cancer. Biochem Biophys Res Commun 2005; 337: 127–32.
Wei Y, Kong B, Song K, Qu X, Jin Q, Yang Q . Involvement of mitochondria-caspase pathway in hemoporfin-mediated cell death. Photochem Photobiol 2007; 83: 1319–24.
Liu F, Gu Y, Liu H, Fu Q, Zhu J, Pan Y, et al. An experimental study on the comparison of photodynamic effects of hematoporphyrin monomethyl ether and hematoporphyrin derivative. Chin J Laser Med Surg 2001; 10: 69–73.
Chen WH, Yu JX, Yao JZ, Shen WD, Liu JF, Xu DY . Pharmacokinetic studies hematoporphyrin monomethyl ether: a new promising drug for photodynamic therapy of tumors. Chin J Laser Med Surg 2000; 9: 105–8.
Du WM, Wang YM, Cheng NN . ADR confirmation and its research methods (2). Chin J Pharmacovigilance 2005; 2: 11–3.
Li P, Sun JG, Huang CR, Xu MJ, Li J, Lu H, et al. Determination of hematoporphyrin monomethyl ether in dogs plasma by HPLC using fluorescence detection. Chin J Laser Med Surg 2005; 14: 212–6.
Yu DK . The contribution of P-glycoprotein to pharmacokinetic drug-drug interactions. J Clin Pharmacol 1999; 39: 1203–11.
Schuetz EG, Furuya KN, Schuetz JD . Interindividual variation in expression of P-glycoprotein in normal human liver and secondary hepatic neoplasms. J Pharmacol Exp Ther 1995; 275: 1011–8.
Acknowledgements
We appreciate all the volunteers participating in this research. We are grateful to the nurses who participated in this study. We appreciate our colleagues X LIU and Q XIANG, who provided editorial assistance.
This study was sponsored by Shanghai Fudan-Zhangjiang Bio-Pharmaceutical Co, Ltd. The work was funded by National Mega Project on Major Drug Development Research (2009ZX09401-001), key project of Science and Technology commission of Shanghai Municipality (064319003).
Author information
Authors and Affiliations
Corresponding authors
Rights and permissions
About this article
Cite this article
Sun, Ph., Zhao, X., Zhou, Y. et al. Tolerance and pharmacokinetics of single-dose intravenous hemoporfin in healthy volunteers. Acta Pharmacol Sin 32, 1549–1554 (2011). https://doi.org/10.1038/aps.2011.132
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/aps.2011.132
Keywords
This article is cited by
-
Hemoporfin-Mediated Photodynamic Therapy for Port-Wine Stains on Extremities
Dermatology and Therapy (2023)
-
Potential role of organic anion transporting polypeptide 1B1 (OATP1B1) in the selective hepatic uptake of hematoporphyrin monomethyl ether isomers
Acta Pharmacologica Sinica (2015)
