
Abstract
Localized surface plasmon resonance (LSPR) of plasmonic nanoparticles and nanostructures has attracted wide attention because the nanoparticles exhibit a strong near-field enhancement through interaction with visible light, enabling subwavelength optics and sensing at the single-molecule level. The extremely fast LSPR decays have raised doubts that such nanoparticles have use in photochemistry and energy storage. Recent studies have demonstrated the capability of such plasmonic systems in producing LSPR-induced hot electrons that are useful in energy conversion and storage when combined with electron-accepting semiconductors. Due to the femtosecond timescale, hot-electron transfer is under intense investigation to promote ongoing applications in photovoltaics and photocatalysis. Concurrently, hot-electron decay results in photothermal responses or plasmonic heating. Importantly, this heating has received renewed interest in photothermal manipulation, despite the developments in optical manipulation using optical forces to move and position nanoparticles and molecules guided by plasmonic nanostructures. To realize plasmonic heating-based manipulation, photothermally generated flows, such as thermophoresis, the Marangoni effect and thermal convection, are exploited. Plasmon-enhanced optical tweezers together with plasmon-induced heating show potential as an ultimate bottom-up method for fabricating nanomaterials. We review recent progress in two fascinating areas: solar energy conversion through interfacial electron transfer in gold-semiconductor composite materials and plasmon-induced nanofabrication.
Similar content being viewed by others

Introduction
Light interacts with the conduction-band electrons of plasmonic nanoparticles (NPs), resonantly exciting coherent oscillations. This phenomenon is known as localized surface plasmon resonance (LSPR) and is the origin of the strong absorption/scattering of incident light by NPs such as those composed of gold (Au NPs). LSPR decays both radiatively and nonradiatively. The former plays a key role in the plasmonic enhancement of the electric field in the near-field regime, whereas the latter is responsible for generating hot electrons with very high kinetic energies, which eventually contribute to particle heating. Hot electrons generated in semiconductor-plasmonic nanostructure (NS) composites have been shown to play a central role in increasing the efficiency of solar energy conversion because of the ability to tune the wavelength over a wide resonance in the visible-to-near-infrared (NIR) region by controlling the physical shape of the NPs.
The decay dynamics of photoexcited Au NPs are well-described by three representative time constants: relaxation from a non-Fermi to Fermi electron distribution through ‘electron–electron (e–e) scattering’ (<100 fs), cooling of hot-electron gas through ‘electron–phonon (e–ph) scattering’ (1–10 ps), and heat dissipation from Au NPs to the environment through ‘phonon–phonon (ph–ph) scattering’ (∼100 ps).1, 2 At the very beginning, plasmon decay occurs through Landau damping on time scales of a few to tens of femtoseconds, generating hot electron–hole pairs.3 Because of their extremely fast deactivation through e–e scattering, only high-potential-energy hot electrons have an opportunity to participate in electron transfer on the Au NP surface, resulting in poor efficiency. Efficient electron transfer, however, can occur even over this short time scale if sufficiently large electronic coupling between the hot-electron and electron-accepting levels is present. This is the case for TiO2, which has a larger density of states in the conduction band, originating from the d-orbital nature, than other typical metal oxides, such as ZnO, SnO2 and In2O3, the conduction bands of which are mostly composed of either s or sp orbitals of the metal atoms. Because of its d-orbital nature, TiO2 is an excellent electron acceptor.4 Indeed, ample ultrafast-spectroscopy studies have been performed, showing that electron transfer from photoexcited dye to the TiO2 conduction band occurs within time scales of 100 fs in dye-sensitized solar cells,5 which is much faster than the excited-state lifetime of dyes.6, 7, 8 Interestingly, as a mechanism of electron transfer from plasmonic metals to semiconductors alternative to hot-electron transfer, chemical interface dumping, in which plasmons decay through a direct electron-separation channel at the interface, has been postulated.9, 10, 11
A decade ago, we realized the potential of Au NPs to act as electron donors to TiO2 NPs when in close contact. Hence, Au NPs can be viewed as photosensitizers instead of commonly employed dyes in dye-sensitized solar cells.12, 13 Au–TiO2 exhibited photocurrent generation upon the excitation of the LSPR band in the presence of appropriate electron donors. Soon thereafter, other work demonstrated plasmon-induced electron transfer from plasmonic NSs composed of Au or Ag to electron-accepting semiconductors, and the concept of plasmon-induced hot-electron transfer gained increasing popularity among the scientific community. A few review papers have examined research activities in this target area: (i) plasmonic-metal NSs that enable the efficient conversion of solar energy to chemical energy through photocatalytic water splitting;14 (ii) plasmon-induced hot-electron generation at NP/metal-oxide interfaces for photovoltaic and photocatalytic devices;15 (iii) the science and technology of plasmon-induced hot-carrier systems, in particular, solar energy conversion process along with hot-electron-induced heating applications toward cancer treatment;16 and (iv) the surface chemistry of Au/TiO2, paying special attention to its behavior in thermal catalysis as well as light-driven catalysis17. Progress in hot-electron transfer research is quite rapid and constantly accumulating. We present an overview of recent progress in plasmonic solar cells and plasmonic photocatalysts, describing the current stage of device performance and material development. In addition, several recent approaches to the dynamics of interfacial hot-electron transfer using ultrafast time-resolved spectroscopy are presented.
In addition to the hot-electron dynamics upon ultrashort pulsed excitation, the steady-state photoexcitation of plasmonic NPs and NSs provides us with innovative techniques. Currently, manipulating—that is, selecting, directing and positioning—NPs, proteins and DNA with external force is applied to fabricate materials at small scales. Optical trapping exerted by a tightly focused laser beam is very powerful for achieving this goal because of its remote and noninvasive nature,18, 19 although other handling techniques using temperature and electrical and magnetic fields have been used. Optical manipulation acts to guide molecules and NPs to intended positions, allowing accumulation or crystallization to ensue. When the plasmonic properties are coupled with conventional optical-trapping techniques, nanoscale manipulations are possible. Such manipulations are not possible with conventional optical-trapping forces, which scale inversely with the cube of the particle diameter.20, 21 Similar to optical forces, the importance of the temperature field created by plasmonic heating has been realized to be an effective means for driving colloids and macromolecules.22 As a secondary topic, we describe the current status of plasmonic-manipulation-based bottom-up fabrication.
Plasmonic hot-electron transfer and plasmon-assisted manipulation have emerged as innovative guidelines for material design and fabrication at the nanoscale. In the following, we examine the current status of the field, highlighting nanomaterial design to achieve high-performance devices. Plasmonic hot-electron transfer has the potential to drastically improve the efficiency and durability of existing solar cells and photocatalysts. Plasmon-assisted manipulation, on the other hand, allows the fabrication of structures rarely achieved by self-assembly or self-organization, realizing advanced functionalities. Together, the two protocols most likely add a new dimension to nanomaterial-based energy devices, nanomedicines and environmental catalysts.
Plasmonic hot-electron transfer
This section describes recent research progress into plasmonic hot-electron transfer. We first show two representative applications, that is, plasmonic solar cells and plasmonic photocatalysts, in which ultrafast hot-electron transfer from a plasmon-excited metal NS to an adjacent semiconductor occurs as the primary process. To understand the mechanism of photoinduced electron transfer in detail, ultrafast spectroscopic techniques are of great use.23 Recent achievements using transient absorption spectroscopy and time-resolved photoemission electron microscopy in the femtosecond and picosecond time regimes are reviewed. Representative devices based on hot-electron transfer presented in this review are summarized in Table 1.
Recent progress in plasmonic solar cells
Plasmonic solar cells using plasmonic metal NS and electron-accepting semiconductors were reported in the pioneering work of the Tatsuma group in 2004 and 2005.12, 13 Tian and coworkers reported that Au NPs can be used as photosensitizers in place of common dye molecules in dye-sensitized solar cells, in which liquid electrolyte, typically the I−/I3− redox pair, is used to shuttle electrons between two electrodes, yielding a nearly 100% incident photon to current conversion efficiency (IPCE) and over 10% power conversion efficiency.24 They demonstrated that the Au–TiO2 system with an appropriate electron donor, in the presence of Fe2+/3+, exhibited sufficiently large photocurrent under excitation of the plasmon band, with a maximum IPCE of 26%. This value is still much lower than that of dye-sensitized solar cells; however, this work strongly impacted the scientific community, given that for Au NPs, the nature of the excitation (collective excitation) is completely different from that of dye (single-electron excitation) and the lifetime of the LSPR is much shorter (~10 fs) than that of excited dye molecules (~1 ns). The authors clearly showed that charge separation occurred through electron transfer from the excited gold particles into TiO2 and successfully applied this phenomenon to visible light to electricity conversion.
Following this work, Misawa and coworkers found that well-controlled gold NSs on a TiO2 single crystal fabricated using electron-beam lithography function as photoelectrodes in aqueous electrolyte.25 The authors concluded that the initial process involves plasmon-induced electron transfer from the gold NSs to the conduction band of TiO2. Interestingly, they could tune the plasmon resonance to the NIR region using a structure with a long axis and found that even 1200-nm light could generate a photocurrent. This work was later extended to plasmon-induced water splitting.26
In the following, recent representative research progress into plasmonic solar cells is presented. Garcia de Arquer et al.27 reported the photoelectric energy conversion of plasmon-generated hot carriers in metal-insulator-semiconductor structures. This structure comprises a complete solid solar cell, where an insulator layer between the Ag metal and the TiO2 semiconductor played an important role in increasing the photovoltage and fill factor (FF) of the device, resulting in the successful improvement of the overall device performance. Figure 1 shows the nanopatterned device structure consisting of FTO (fluorine-doped thin oxide)–TiO2–Al2O3–Ag layers. The Al2O3 insulator is used to block back electron transfer from TiO2 to Ag. The dependence of various solar cell parameters on the Al2O3 thickness (δ) was examined. Whereas the short-circuit current gradually decays as the Al2O3 thickness increases from 0 to 4 nm, the open-circuit voltage (VOC) and the FF increase for thicknesses between 0 and 0.5 nm. In consequence, the power conversion efficiency (PCE) reaches 0.03% for the champion device in the report. The authors further concluded, after comparison of the data, that electrons derived from Ag LSPR are emitted over the barrier, whereas Au LSPR electrons undergo Fowler–Nordheim tunneling through the barrier in the FTO-TiO2-Al2O3-Au device structure.

Nanopatterned device structure consists of FTO-TiO2-Al2O3-Ag layers: (a) schematic image. (b) The effect of the interface on the junction properties. LSPR excited by an incoming photon can result in the creation of an electron-hole pair, which can be split by the built-in field in the interface. Passivation of interface states prevents pinning. Depending on their energy, the excited electrons can either tunnel to the TiO2 or undergo Schottky emission over the barrier. The height of the barrier determines the difference of metal work function (Wf) and insulator electron affinity (χins). Adapted with permission from Garcia de Arquer et al.27 (Copyright 2013 American Chemical Society).
Mubeen et al.28 reported a stable, wholly plasmonic photovoltaic device in which photon absorption and carrier generation occur exclusively in the plasmonic metal. The authors used a metal (Au-nanorod)–semiconductor (TiO2)–metal (Ti) structure (Figure 2). A field established at the metal–semiconductor Schottky junction can separate charges. The negative carriers are high-energy (hot) electrons produced immediately following plasmon dephasing in the Au nanorods. The authors explain that the carriers are energetic enough to clear the Schottky barrier or quantum mechanically tunnel through it, thereby producing the output photocurrent. The effect of TiO2 thickness was examined. The measured VOC rapidly increased when the TiO2 thickness increased from 10 to 50 nm. Devices fabricated with 50-nm TiO2 layers had a VOC as high as 210 mV, a short-circuit current density of 26 μA cm−2 and a FF of 0.3. Under short-circuit conditions, an IPCE of ∼1.0% and IQE of 2.75% were found, which are considerably higher than those previously reported for plasmonic photovoltaics under zero-bias conditions. Interestingly, the cells showed marked stability in accelerated aging tests (up to 1 month).

Structure and hot carrier transfer process in the plasmonic photovoltaic device. The oriented gold nanorod arrays are coated TiO2 using atomic layer desorption (ALD). Ti metal (whose work function is less than that of Au) serves as the back contact. Adapted with permission from Mubeen et al.28 (Copyright 2014 American Chemical Society).
Very recently, Nakamura et al.29 reported another example of a solid-state plasmonic solar cell using a TiO2/NiO p–n junction with Au NPs. The reported solar cell had a 0.56 V VOC, 108 μA cm−2 JSC and 0.55 FF, corresponding to a 0.033% power conversion efficiency. The annealing procedure further increased the VOC to 0.73 V because of the firm adhesion between TiO2 and NiO. The solar cells also achieved long-term stability after irradiation for three days.
So far, liquid-junction solar cells seem to have an advantage in terms of high solar-cell performance compared with solid-state devices. Lee and coworkers reported that when illuminated with simulated sunlight, the device (Au–TiO2/V3+(0.018 M), V2+(0.182 M)/Pt) reproducibly and sustainably produced a VOC of 0.50 V and a JSC of 0.5 mA cm−2, corresponding to a power conversion efficiency of 0.095%.30 They used a V2+/V3+ redox couple and TiO2-coated Au arrays. The appropriate choice of redox couple and broadband light absorber properties of gold nano-islands enabled the high VOC and JSC, respectively. A typical set of photovoltaic results yielded VOC =478 mV, JSC=470 mA cm−2, FF=0.423 and PCE=0.095%. The device showed good stability for more than 1000 h under ambient conditions.
Here, as a final example, a recent interesting study obtained a high solar-cell power conversion efficiency by combining the Au-TiO2 system with a perovskite methyl-ammonium lead iodide (MAPbI3) solar cell.31 Mali and coworkers reported in situ-processed gold NP-embedded TiO2 nanofibers that enabled plasmonic perovskite solar cells to exceed 14% power conversion efficiency.32 In the device structure (Figure 3), both MAPbI3 and Au NPs work as visible-light absorbers, and electron transfer occurs from both sensitizers to the TiO2 conduction band. The authors concluded that the observed increase in current density may be attributed not only to an increase in the excitation of the perovskite by the effects of LSPR associated with near-field enhancement and scattering but also to the generation of additional photocurrent arising from the LSPR-induced direct transfer of hot electrons from plasmonic structures to the conduction band of TiO2. The solar-cell performance depends on the weight ratio of Au NPs to TiO2; the best power conversion efficiency of 14.37% was obtained with 0.3 wt%.

Top and cross-sectional FESEM (field emission scanning electron microscopy) images, image and energy level diagram of nanofiber-based perovskite solar cells. (a) FESEM cross-sectional image of fabricated perovskite solar cell. (b) Higher-magnification image of selected area of perovskite with Au@TiO2 nanofibers. The inset shows a TEM (transmission electron microscopy) image of perovskite covered with Au@TiO2 nanofibers. (c) Schematic of device architecture. (d) Schematic of energy levels for TiO2, Au nanoparticle, CH3NH3PbI3, spiro-MeOTAD and gold contact. (e) Image of final perovskite solid-state device. Adapted with permission from Mali et al.32. (Copyright 2016 The Royal Society of Chemistry).
Recent progress in plasmonic photocatalysts
Ever since the discovery of the Honda–Fujishima effect,33 where the combination of a TiO2 photoanode and Pt cathode was reported to split water under photoirradiation, TiO2 has become the most popular and extensively studied photocatalyst. However, the limited range of photoabsorption in the UV region (less than ~380 nm) makes this material difficult to use for solar energy conversion as the UV band only comprises a small portion of sunlight (<5%). Thus, great effort has been made in developing visible-light-responsive semiconductor photocatalysts by engineering the band structures of transition metal oxides, nitrides and oxynitrides.34
In an attractive way to obtain a visible light (or even NIR-light) response in TiO2–water splitting, Misawa’s group extensively studied plasmon-enhanced photoelectric conversion and water oxidation via visible- and NIR-light irradiation by employing TiO2 single-crystal photoelectrodes, in which gold nanorods are arrayed on the surface. In the study, a potassium perchlorate aqueous solution was employed as an electrolyte solution without additional electron donors; thus, they concluded that water molecules provided the electrons. The nanorods exhibited strong optical absorption transition due to the longitudinal mode along the ~200-nm-long axis of the nanorod in the NIR region (around the 1050-nm wavelength); with the excitation of the absorption band, the electron transfer produced nearly 10% IPCE in their photoelectrochemical cell. The details of this and related studies are summarized in review papers from Misawa’s group, with discussions on how to optimize the materials and physical structures with semiconductor photoelectrodes for plasmon-induced water splitting.35, 36 Some very recent work from other groups is presented here.
DuChene and coworkers reported prolonged electron lifetimes in plasmonic-metal/semiconductor heterostructures and tested solar photocatalysis to produce H2 from water.37 Figures 4a–d shows the structure of the Au/TiO2 heterojunctions. Au NPs (d=20±5 nm) were photochemically grafted directly onto the TiO2 nanowires to construct Au/TiO2 heterojunctions. Scanning electron microscopy (SEM) analysis revealed that the Au NPs were distributed along the TiO2 support but were predominantly located at the nanowire terminus. They discovered that the electrons transferred from Au to TiO2 under visible-light irradiation displayed excited-state lifetimes two-orders-of-magnitude longer (of the order ~103 s) than those of electrons photogenerated directly within TiO2 via UV excitations. The dimensions of the semiconductor scaffold used in their study (0.2–2 μm) are suitable in supporting a depletion layer within the TiO2 nanowire. The prolonged lifetimes likely result from the significant Schottky barrier (with a height of ~1.0 eV) established at the Au/TiO2 interface. The depletion layer within the semiconductor promotes charge separation by sweeping these hot electrons away from the Au/TiO2 interface. Recombination is further suppressed by the absence of valence band holes in TiO2 under visible light irradiation. Hence, efficient H2 evolution from the Pt wire cathode in the presence of methanol as a sacrificial reagent was inferred.

Structural analysis of a typical plasmonic photoanode. (a) SEM image of a plasmonic Au/TiO2 photoanode on the FTO glass substrate. (b) HAADF-STEM (high-angle annular dark-field scanning transmission electron microscopy) image demonstrating direct physical contact at the Au/TiO2 interface (dAu(111)=0.235 nm). Adapted with permission from DuChene et al.37. (Copyright 2014 Wiley-VCH Verlag GmbH and Co. KGaA, Weinheim) (c) Experimental and simulated extinction spectra of the Au nanocages (marked NCs in the figure) NCs and TiO2–Au NCs. (d) Photocurrent spectra of the Au NCs and porous TiO2 electrode under visible-light illumination. The simulated extinction spectrum for the Au NCs (shown in c) is presented for comparison. (e) Photoluminescence (PL) spectrum of the TiO2 electrode (red). The photocurrent- and the PL spectra are shown in the same width of photon energy. The PL peak at 3.35 and 3.18 eV, which correspond to the interband transition of TiO2 and to the PL from the SnO2 substrate, respectively, are highlighted in gray. Photocurrent spectrum of the Au NCs–TiO2 (shown in d) is presented for comparison (blue). (f) Model of the LSPR-mediated hot-electron transfer across the TiO2/Au interface. Adapted with permission from Kodiyath et al.38. (Copyright 2014 The Royal Society of Chemistry).
Kodiyath et al.38 reported the visible light photodecomposition of acetaldehyde by TiO2-coated gold nanocages. They found that plasmon-mediated hot-electron transport occurred via defect states, resulting in reactivity both in the visible and NIR regions. Skeletal gold nanocages were prepared and coated with amorphous TiO2 layers via a wet-chemistry route and exhibited enhanced visible light photodecomposition activity toward acetaldehyde (one of the most harmful volatile organic compounds). Theoretical simulations elucidated that the electromagnetic field was enhanced over the visible-light-illuminated Au nanocages through LSPR, forming a number of hot spots on the surface. Figures 4c–f shows the experimental and simulated extinction spectra of the Au nanocages and TiO2–Au nanocages. The peaks in the photocurrent spectra of the Au nanocages and the porous TiO2 electrode under visible-to-NIR-light illumination were much sharper than those in the simulated extinction spectrum, indicating that the photocharges are transported via a certain discrete state at the interface. The photoluminescence (PL) spectrum at 10 K of the porous TiO2 electrode showed a couple of peaks due to defect states. The sharp photocurrent peaks are attributed to hot-electron transport via the defect states of TiO2 (see schematic in Figure 4f).
Nanosheets were combined with a Au/TiO2 system to improve the photocatalytic properties. Ide and coworkers found that the activity of Au NP-loaded P25 TiO2 (Au/P25) photocatalysts was enhanced threefold by simply mixing Au/P25 with photocatalytically inactive hexagonal boron nitride (h-BN) insulator nanosheets.39 The activity was evaluated by the oxidative decomposition of formic acid in water under visible light irradiation. The authors attributed the effect to electron transfer from photoexcited Au/TiO2 to the h-BN nanosheets and the retardation of charge recombination.
Mechanism revealed by ultrafast spectroscopy
The first direct observation of plasmon-induced electron transfer from metal NPs to a semiconductor was reported in 2007 by Furube et al.40 They measured the femtosecond transient absorption at an IR wavelength following visible excitation at the plasmon resonance of Au NPs physically attached to a TiO2 surface. The IR transient absorption signals due to injected conductive electrons in the conduction band of TiO2 were detected and found to give an electron transfer time of below 220 fs and a reaction yield of approximately 40%. This result suggested that this transfer proceeds in competition with e–e scattering in the metal, as mentioned in the introduction. No further electron transfer was observed after the e–e scattering relaxation within ~100 fs, in which the electron gas was thermalized according to a Fermi distribution. Therefore, only hot electrons in Au NPs can participate in electron transfer to TiO2. Further investigations using different excitation wavelengths around the plasmon resonance suggested that the direct generation of electron–hole pairs through interband transitions (from the d band to the sp band) at a shorter wavelength than the plasmon peak was more beneficial for a higher electron-transfer yield. In fact, a nearly 100% yield was obtained when a 480-nm wavelength laser was used for excitation. Additional discussions related to electric-field enhancement occurring in fine NSs are presented in a previous review article.41 Further discussion might be possible as follows: an interband transition generates electrons excited from the d band of gold to the sp band, leading to the generation of very hot holes accompanied by less-energetic hot electrons.16 The hot holes may be filled by sp-band electrons via an Auger mechanism, resulting in very hot electrons, which can transfer to TiO2.
A back electron transfer mechanism from the semiconductor to the original metal NPs has also been reported.40, 42 The transient absorption signals from the electrons in TiO2 decayed over picosecond timescales, and the amplitude at a 1-ns time decay was less than 10% of the initial intensity, indicating that most of the injected electrons disappear through recombination. This is a significant drawback for applications toward solar energy conversion, where long-lived electrons in the semiconductor are required to generate a photocurrent or to initiate reductive chemical reactions at the surface. Du and coworkers showed that the electron lifetime in a TiO2 NP after plasmon-induced electron injection from a Au NP increased from 8 to 128 ps when the diameter of the TiO2 NP was extended from 9 to 50 nm. They concluded that the TiO2 provided a larger volume through which electrons could diffuse until back electron transfer became dominant.42
Within a few years of these primary reports, other work on the dynamical aspects of plasmon-induced hot-electron transfer between metal NSs and semiconductors was published. In the following, this work is discussed in chronological order of publication. Cushing et al.43 investigated the plasmonic energy transfer mechanism in Au@SiO2@Cu2O and Au@Cu2O sandwich-structured NSs using transient absorption and photocatalysis action spectral measurements. In addition to energy transfer, they observed minor contributions to electron transfer from Au to Cu2O for Au@Cu2O. They reported that the gold core converted the energy of incident photons into LSPR oscillations and transferred the plasmonic energy to the Cu2O semiconductor shell via resonant energy transfer, which generated electron–hole pairs in the semiconductor via the dipole–dipole interaction between the plasmonic metal and semiconductor. Figure 5 shows the transient absorption data and a schematic comparing the kinetics between the two NSs, Au@SiO2@Cu2O and Au@Cu2O. The transient absorption signal probed at 800 nm is from free-carrier absorption in the conduction band of Cu2O. The rise time of the signals from Au@Cu2O has fast (<100 fs) and slow (~2 ps) components. The fast component is most likely the result of direct electron-transfer interactions. In contrast, for Au@SiO2@Cu2O, only the slower rise-time component survives because the energy transfer scheme is more favorable than that expected for Au@Cu2O with the insulating layer.

Ultrafast pump−probe measurements for core-shell and sandwich structures. (a) Transient absorption for Au@Cu2O and Au@SiO2@Cu2O nanostructures acquired with a wavelength of 650 nm. Decays are fit showing nearly identical recombination rates. (b) Schematic representation of the various transfer mechanisms that can occur in the Au@Cu2O structure. Also shown in the diagram are the pump, probe (free-carrier absorption), and recombination paths. Adapted with permission from Cushing et al.43. (Copyright 2012 American Chemical Society).
Tang and coworkers investigated the photocatalytic property of Ag@AgCl cubic cages, that is, Ag NPs encapsulated in an AgCl cubic cage.44 They detected an ultrafast electron-transfer process (⩽150 fs) from Ag NPs to the AgCl surface, which facilitated the charge separation efficiency in this system, contributing to the high photocatalytic activity and stability of the Ag@AgCl photocatalyst against organic dye degradation. Figures 6a–d shows the 470-nm transient absorption kinetics of Ag NPs as well as Ag NPs coupled with AgCl NPs after 400-nm excitation. For Ag NPs only, the fast decay of the photon-induced electron–plasmon bleaching (τ1≈1.8 ps) was dominated by electron–electron scattering and electron–phonon scattering (energy transfer from the hot electrons to the lattice). The slow recovery of the transient photon-induced absorption (τ2>2 ns) to equilibrium was attributed to heat dissipation from the Ag NPs to the surrounding matrix (that is, phonon–phonon interaction). For the Ag@AgCl nanohybrid system under the same experimental conditions, the absence of electron–plasmon bleaching and hot-electron cooling indicated strong electronic coupling between the Ag and AgCl conduction bands. The authors attributed this to the efficient injection of photoinduced energetic electrons from the Ag NPs to AgCl. The injection time was far shorter than the transient absorption time resolution (150 fs). From these observations, the authors proposed a mechanism (Figure 6f). Upon light irradiation, electromagnetic waves are highly confined and amplified at certain interfacial hot spots (red spots in the inset of Figure 6f). The excited surface plasmons rapidly decay, producing many hot electrons, which transiently occupy empty states in the conduction band of silver above the Fermi energy. A significant fraction of the excited electrons is transferred to AgCl. The authors suggested that the holes from silver oxidize organic dyes (methyl orange).

Comparisons of 470-nm transient absorption kinetics of (a, b) Ag nanoparticles and (c, d) Ag nanoparticles coupled with AgCl nanoparticles after 400-nm excitation. (e) Schematic illustration of the ultrafast electron transfer from Ag to AgCl after excitation. (f) The proposed mechanism of photocatalytic organic dye degradation for the Ag/AgCl hybrid structures. Adapted with permission from Tang et al.44. (Copyright 2013 WILEY-VCH Verlag GmbH and Co. KGaA, Weinheim).
Bian and coworkers reported that the lifetime of electrons injected in the TiO2 nanostructure from Au NPs can be influenced by the physical shape of the Au/TiO2 superstructure.45 To the best of our knowledge, they are the second group that has directly observed electron transfer from plasmon-excited Au to TiO2 using an ultrafast time-resolved experiment. They synthesized plasmonic photocatalysts by modifying TiO2 mesocrystals (micrometer plate-like crystals) with Au NPs via simple impregnation. The Au NP sensitizers show strong photoelectrochemical reactivity in the visible-light region due to LSPR. The authors conducted transient absorption spectroscopy and demonstrated that a substantial number of electrons, injected from the Au NPs to the TiO2 mesocrystals, directionally migrated from the basal surfaces to the edges of the plate-like mesocrystals through the TiO2 nanocrystal network and are temporally stored there for further reaction. Such electron flow significantly retarded the charge recombination of these electrons with the holes from Au NPs.
Figures 7a and b indicate the apparent quantum efficiency and SPR spectra of the Au/MesoTiO2 system for the photocatalytic degradation of methylene blue and its absorption spectrum in the visible region. Significantly faster degradation was observed for Au/MesoTiO2 compared with four other references: MesoTiO2 only, Au/MesoTiO2-PD (photochemical deposition), Au/Nano-TiO2 and Au/P25 (AEROXIDE TiO2) systems. In the Au/MesoTiO2 system, Au NPs tend to exist at the edge of the TiO2 plate-like mesocrystals, whereas in the Au/MesoTiO2-PD system Au NPs tend to exist on the large surface of the TiO2 plate-like mesocrystals. The difference in photocatalytic response mechanisms was clarified by the transient absorption kinetics (Figures 7c and d), in which he transient absorption traces observed at 1000 nm for the Au/MesoTiO2, Au/MesoTiO2-PD and Au/P25 systems are plotted. Note that more than 60% of electrons in the Au/MesoTiO2 system have prolonged electron lifetimes of ~10 ns, whereas electrons in the Au/MesoTiO2-PD and Au/P25 TiO2 systems recombine within a few nanoseconds. The authors concluded that the electrons injected from the excited Au NPs on the basal surfaces of MesoTiO2 are effectively delivered to the lateral surfaces of the crystal through the TiO2 nanocrystal network. This significantly retards charge recombination, thus enhancing the visible-light photocatalytic performance associated with the degradation of organics.

(a) Apparent quantum efficiency (AQE) and absorption spectra of the Au/MesoTiO2 system in photocatalytic degradation of methylene blue (MB) and visible absorption spectrum of MB. (b) Kinetic linear fitting curves for liquid-phase photocatalytic degradation of MB over different samples under visible-light irradiation (460−700 nm). (c) Time-resolved diffuse reflectance spectra observed after 530-nm laser flash photolysis of Au/MesoTiO2 in ambient air. (d) Normalized transient absorption traces observed at 1000 nm for Au/MesoTiO2, Au/MesoTiO2-PD, and Au/P25 systems. Adapted with permission from Bian et al.45 (Copyright 2013 American Chemical Society).
Strong interactions in nano-heterostructures composed of Au NPs and quantum dots have been reported by Wu and coworkers. They studied plasmon-induced hot-electron transfer from the Au tip to the CdS rod in CdS–Au nano-heterostructures using femtosecond transient absorption spectroscopy.46 The optical excitation of plasmons in the Au tip leads to hot-electron injection into the CdS rod with a quantum yield of 2.75%. They measured the transient absorption spectra and kinetics of the CdS–Au nanorods after 590-nm wavelength excitation (Figure 8). The control 2 sample was composed of a mixture of CdS nanorods and Au NPs. In the visible region, the spectra were dominated by heating-induced Au LSPR spectral changes (bleaching around the LSPR peak and positive wings on the both sides of the original LSPR band because of band broadening). Comparing the kinetics probed at 452 nm of the CdS–Au and control 2 samples (Figure 8b), a notable difference is found, which indicates that bleaching occurred for the CdS 1Σ band in CdS–Au. The direct observation of intraband absorption of the 1σ electron at 3000 nm proves that hot-electron transfer from Au to the CdS nanorods occurs. Specifically, intraband absorption was not observed in the control 2 sample (Figure 8c); however, in the CdS–Au nanorods sample, an intraband signal was clearly observed. The signal rise time was 0.097 ps, and the multi-exponential decay had a half-life of 1.83 ps. Based on a comparison of the sizes of the transient absorption signals at 590- and 400-nm excitation, the authors estimated the quantum yield of hot-electron transfer to be ~2.75%. Interestingly, they compared the hot-electron injection yield and charge recombination times of CdS–Au nanorod systems with a previous report on Au-sensitized TiO2 NPs, in which the plasmon-induced injection yield was measured at ~40% and the recombination time was hundreds of picoseconds. The high injection yield in Au/TiO2 was attributed to a fast electron injection rate, resulting from a high density of electron-accepting states in TiO2. The relatively slow recombination in Au/TiO2 is attributed to electron diffusion within the TiO2 NPs.

Transient absorption spectra and kinetics of CdS-Au nanorods after 590-nm excitation. (a) Transient absorption spectra of CdS-Au nanorods (upper panel) and control 2 (lower panel) at indicated delay time windows (from 0.2 to 1000 ps). Control 2 is a mixture of CdS nanorods and Au NPs in which the nanorod concentration and the sample absorbance at 590 nm are the same as the CdS-Au nanorod sample. (b) Kinetics probed at 452 nm for CdS-Au nanorods (black solid line) and control 2 (red dashed line). (c) Kinetics probed at 3000 nm for CdS-Au nanorods (red circles) and control 2 (gay dashed line). The black solid line is a fit to the kinetics of CdS-Au. Adapted with permission from Wu et al.46 (Copyright 2013 American Chemical Society).
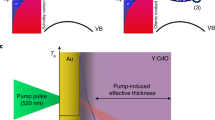
Subsequently, the same group reported efficient hot-electron transfer by a plasmon-induced interfacial charge-transfer transition (PICTT) in a system of CdSe–Au nano-heterostructures with a Au tip and CdS rod.10 They proposed the PICTT mechanism, which is different from conventional plasmon-induced hot-electron transfer (PHET) or direct metal-to-semiconductor interfacial charge-transfer transition (DICTT). In PICTT, the metal plasmon serves as a light absorber, but strong interdomain coupling and mixing of the metal and semiconductor levels lead to a new plasmon decay pathway—the direct generation of an electron in the semiconductor and an electron–hole pair in the metal without the generation of hot electrons (Figure 9). Through transient absorption experiments, the gold plasmon appeared to be strongly damped by CdSe through interfacial electron transfer. The quantum efficiency of PICTT was found to be higher than 24%.

Metal-to-semiconductor charge-separation pathways. (a) Conventional PHET mechanism, in which a photoexcited plasmon (SP, blue ellipsoid) in the metal decays into a hot electron–hole pair (solid and open red circles in the dotted ellipsoids) through Landau damping, followed by injection of the hot electron into the CB (conduction band) of the semiconductor. The electron–hole pair has a broad distribution of initial electron and hole energies; only two are shown for clarity. (b) Optical excitation of an electron in the metal directly into the CB of the semiconductor through the DICTT pathway. (c) The newly demonstrated PICTT pathway, where the plasmon decays by directly creating an electron in the CB of the semiconductor and a hole in the metal. VB is the semiconductor valence band and hv indicates the excitation photons. Adapted with permission from Wu et al.10 (Copyright 2015 AAAS).
Three interesting transient absorption studies have been reported in the past few years on plasmon-induced electron transfer from Au NSs to TiO2 in well-controlled structures. First, Harutyunyan et al.47 reported anomalous ultrafast dynamics of plasmonic hot electrons in hybrid metal/oxide NSs with hot spots when they varied the geometry and composition of the NS and the excitation wavelength. They showed a large ultrafast contribution to the excited electron decay signal in the hybrid NSs of gold nanodisks with diameters of 100–150 nm on top of a 30-nm-thick continuous gold film separated by a spacer layer (several nanometers thick) containing hot spots. Based on theoretical models, the effect was attributed to the generation of plasmonic hot electrons from hot spots within 45 fs. When TiO2 was used as a spacer layer, they observed nonlinear transient bleaching, which was explained by the decrease in electron density in gold due to the injection of electrons into the TiO2 spacer layer.
Second, Kumar and coworkers reported the ultrafast and efficient transport of plasmonic hot electrons employing graphene from photoexcited AuNPs to cause hydrogen evolution.48 This system represented a Pt-free, highly efficient, visible light responsive photocatalyst. Figures 10a–c present a schematic of graphene-coated Au NPs for CO2 photoconversion. The authors also measured the transient absorption by employing a 575-nm pump and an IR probe for these electron-transfer systems with TiO2 acceptors. The experimental result (Figures 10d and e) showed more efficient hot-electron transfer in r-GO-AuNPs than in GO-AuNPs or AuNPs (here, GO denotes graphene oxide, and r-GO its reduced form). This trend is in good agreement with the experimental results of photocatalytic reactions yielding formic acid, highlighting the role of the graphene layer in effecting efficient photocatalytic reactions.

Schematic of the graphene-coated gold nanoparticle for CO2 photoconversion. (a) HR-TEM image. (b) Energy dispersive X-ray mapping image of r-GO-AuNPs (Au (blue), carbon (red)). (c) Schematic representation of hot-electron generation and the role of graphene layer as an efficient electron acceptor and transporter to generate hydrogen and subsequent conversion of CO2 into formic acid (HCOOH) and methanol (CH3OH). Adapted with permission from Kumar et al.48. (Copyright 2016 American Chemical Society) (d) Experimental scheme of femtosecond transient absorption spectroscopic analysis. (e) Transient absorption kinetics at 2978 cm−1 of TiO2 nanoparticle in the presence of Au NPs (blue), GO- Au NPs (red), and r-GO-Au NPs (black). Adapted with permission from Kumar et al.48 (Copyright 2016 American Chemical Society).
Third, Zeng et al.49 showed that strong light–matter coupling between confined photons on a semiconductor waveguide and localized plasmon resonances on metal nanowires can modify the efficiency of the photoinduced charge transfer rate of plasmonic-derived (hot) electrons into accepting states in the semiconductor material. The structure of the sample and a diagram illustrating the formation of plasmon−waveguide hybrid states are shown in Figures 11a–c. Pump−probe transient absorption experiments were performed by pumping electrons into the P+ states (Figures 11a–c) and probing the transient occupation of electrons in the conduction band of TiO2. The authors tested for effects resulting from the manner of contact (Figures 11d and e). For a glass substrate, electron injection from Au nanowires is unlikely, as the energetics are unfavorable, whereas electron injection is possible with Ohmic contact—the lifetime of the charge-separated states is shorter than that at the Schottky contacts, in which the energetic barrier prevents the injected electrons from recombining with holes. The actual data showed efficient hot-electron transfer for a sample with a Schottky barrier compared with two other samples. The authors also found that the electron-injection efficiency depends on the grating period, demonstrating that modifying the electro-magnetic environment of plasmonic systems can modify the rate and efficiency of plasmonic hot-electron injection.

Strong waveguide−plasmon interactions. (a) Au gratings of 80 nm in width and 30 nm in height were deposited on top of 200-nm films of TiO2. The film can support waveguide modes (red wavy lines) which when resonant with the localized plasmon resonances of the wires can lead to the formation of strongly coupled waveguide−plasmon polaritons. The excitation of these polaritons depends on the periodicity p of the nanowire grating, for normally incident light. (b) SEM image of lithographically-made nanowires, the scale bar shown corresponds to 500 nm. (c) Diagram illustrating the formation of plasmon−waveguide hybrid states (P±, split in energy by ħΩ) from uncoupled waveguide and plasmon states characterized by having energies EW and EP, respectively. The pump−probe experiments were carried out by pumping into P+ states and probing the transient occupation of electrons in the conduction band of TiO2 (represented by the curve vertically shifted by Eg from the valence band). (d) Electron injection. Diagrams representing the differences in the electrical nature of the contacts of E: On an insulating substrate, electron injection is not readily possible. On Ohmic contacts electron back transfer from semiconductor to metal can take place limiting the lifetime of the charge-separated state. On Schottky contacts the barrier slows down this back reaction. (e) Transient spectra for Au nanowire gratings deposited directly on thin films of TiO2 forming a Schottky contact, deposited with a 2-nm Ti adhesion layer, leading to an Ohmic contact and on a glass (insulator) substrate. Adapted with permission from Zeng et al.49 (Copyright 2016 American Chemical Society).
Mechanism revealed by ultrafast PEEM
Time-resolved multi-photon photoemission electron microscopy (TR-MP-PEEM)50 is a powerful method to further investigate the mechanism of plasmon-induced hot-electron transfer from plasmonic metal structures, as this method has a time resolution of less than 10 fs and a spatial resolution below 10 nm. Sun et al.51 successfully applied this technique to the direct imaging of the near-field and dynamics of SPR on gold NSs. Various kinds of gold NS arrays (such as rods, nanodisks and their dimers) fabricated by electron-beam lithography were investigated via excitation with femtosecond NIR laser beams. When the LSPR bands overlapped with the spectrum of the femtosecond laser, a plasmon was efficiently excited and promoted multi-photon emission. The PEEM images featured a local near-field distribution of the LSPR on different Au NSs. In addition, the LSPR dynamics were studied by an interferometric time-resolved pump-probe technique combined with PEEM. This method provides detailed information on the oscillation and dephasing of the LSPR field. Figures 12a–d shows an SEM image of a Au nanorod array on Nb-doped TiO2. The PEEM images of the same array excited using light from a mercury lamp and p-polarized NIR femtosecond laser pulses are clearly different. The latter produced hot spots at the four corners of each rod, as photoemission can occur via a multi-photon process at the location at which the near-field intensity is highly concentrated. The dephasing time of the plasmon oscillation was evaluated to be less than 15 fs, suggesting that hot-electron generation occurs over such timescales at these hot spots.

(a) SEM image of an Au nanorod (95 × 3180 × 340 nm) array on Nb-doped TiO2. (b) A sketch map of the irradiation setup for the PEEM measurements. PEEM images of the same array excited using (c) a mercury lamp and (d) p-polarized NIR femtosecond laser pulses in which hot spots can be observed at four corners of each rod. A dashed line rectangle in c and d indicates the outline of an Au nanorod. The light was irradiated from the left side at an incident angle of 74°. The wave vector in the plane of the sample surface k// is indicated, and for the p-polarized beam used, the polarization in the plane of the sample surface E// is parallel to k//. Adapted with permission from Sun et al.51. (Copyright 2013 NPG) Time-resolved PEEM measurements. (e) Schematic diagram of the setup for the time-resolved PEEM measurements. (f) Evolution of the PE intensity for both p- and s-polarized light excitation (corresponding to the dipole and quadrupole LSPR modes, respectively) within the phase delay of (0−20) × 2π rad (corresponding to the delay time of 0−56 fs). The inset in f shows the time-resolved PE signals expended in the phase delay within (2−6) × 2π rad. PEEM measured and numerical simulated PE intensity for the dipole mode (g) and the quadrupole mode (h) as a function of the delay time between pump and probe pulses. Careful analysis and comparison of the PEEM experimental data with calculations yield a dephasing time of 5 and 9 fs for the dipole and the quadrupole mode, respectively. Adapted with permission from Ref. 52. (Copyright 2016 American Chemical Society). NIR near-infrared; PEEM, photoemission electron microscopy; SEM, scanning electron microscopy.
Very recently, the same group reported a more detailed study on the few-femtosecond dephasing times of 5 fs for the dipole mode and 9 fs for the quadrupole mode in Au NPs (Figures 12e–h).52 These studies provide very important information on the location and timescale of hot-electron generation, which will benefit the future design of plasmonic solar cells based on the assumption that electron-accepting materials must be properly placed to electronically interact with hot electrons.
Optically driven plasmonic nanofabrication
This section describes the fabrication of optically driven materials using plasmonic NPs and NSs. To examine the mechanism of nanofabrication, we must consider micro- and nanoscale events, rather than the bulk-scale synthesis. We focus on events trapped on substrates rather than those occurring in bulk solution because structures fabricated on substrates are likely to find future device applications. Moreover, plasmonic NPs and NSs fabricated on transparent substrates enable in situ optical characterization and postmortem analyses by SEM and atomic force microscopy (AFM). Table 2 summarizes the driving forces and platforms associated with design paradigms. We divided the driving force of light-induced nanoarchitecture formation into three categories.
First, optical forces can be used in laser/optical trapping or optical tweezers.53, 54 The concept of optical trapping is given in Scheme 1. The primary force in these manipulations is a gradient force that moves a particle/molecule to the focal position and maintains the particle at that position (Scheme 1, left). Micrometer- and submicrometer-sized colloidal particles, bacteria and cells can be optically trapped. Because of the larger polarizability of metals, plasmonic NPs are more stably trapped than other dielectric particles of similar size. For light-absorbing plasmonic NPs, an additional scattering force pushes the particle along the propagating direction of the beam (Scheme 1, center). This scattering force is not helpful in 3D trapping but is useful for the 2D trapping of particles against a wall. Furthermore, incorporating plasmonic substrates (Scheme 1, right) augments the optical force through the plasmon-enhanced light field.
Optical forces can be strongly enhanced when coupled with plasmonic NSs; this technique is called ‘plasmon-assisted’ or ‘plasmon-enhanced’ optical trapping.55, 56 Using the plasmon-assisted approach, one can manipulate single Au NPs, protein molecules and amino-acid clusters with submicrometer precision. Optical forces provide a powerful bottom-up approach to material fabrication.
Second, plasmonic heating or photothermal effects arising from nonradiative LSPR decay is a major pathway of energy release of absorbed light via plasmonic NPs.3, 57 As a result of heating confined to a local area, various processes can evolve. Thermophoresis or thermodiffusion is the migration of solutes and solvents from hot to cold or vice versa triggered by temperature gradients. The thermodiffusion coefficient DT is usually scaled by a common diffusion coefficient D, yielding the so-called Soret coefficient ST=DT/D.58 Here, a positive DT occurs for thermophobic solutes that move from hot to cold, whereas a negative DT is assigned to thermophilic solutes that tend to move from cold to hot. Scheme 2 shows an example of thermophoretic DNA trapping assisted by thermal convection in an aqueous solution confined in a microscale chamber, which is heated internally by illumination with an IR laser beam (λ=1480 nm), which is absorbed by water.58 DNA accumulation occurs at the lower surface (shaded areas) of the chamber near the hot spot (contoured area).
When plasmonic NPs or NSs are illuminated, a temperature gradient is formed in solution. In addition to buoyancy-driven thermal convection, Marangoni convection can occur as a result of surface tension gradients generated by temperature gradients formed at interfaces.59 These processes contribute to the transport of molecules and small particles, resulting in accumulation and crystallization under appropriate conditions. Apart from this migration or flow, microbubbles can form if Au NPs and NSs are strongly heated in a liquid environment. For isolated Au NPs, bubbles are produced from superheated water at particle temperatures of 500–550 K.60 The ability of microbubbles to collect and displace molecules and colloids provides an opportunity to assemble small-scale structures if their positions are properly controlled.61
Third, we refer to the action of a strongly enhanced electric field formed around plasmonic NPs under photoillumination through two-photon or multi-photon absorption, resulting in designed structures.62 Remarkably, high-power femtosecond lasers produce unusual geometrical constructions, which we highlight below. Importantly, the above three major driving forces for fabrication are always interconnected and may not necessarily arise separately.
As for the platform that couples with light and promotes the above driving forces, precisely designed plasmonic NSs prepared by the electron-beam lithography/lift-off technique or nanosphere lithography are preferred because well-defined structures must be fabricated reliably and reproducibly regardless of location on the substrates.63, 64 However, nano-islands65 prepared by vacuum evaporation followed by thermal annealing are frequently used as nanostructures with proven advantages in nano-manufacturing applications for their cost-effective and time-saving fabrication. As described below, a simple single plasmonic NP can constitute a platform for nanofabrication. When illuminated with a focused laser beam, a single Au NP experiences both optical forces and photothermal effects. We review a number of example structures that have been fabricated.
Optical force-based fabrication
Figure 13, depicts the plasmonic NS-enhanced optical force-based fabrication of a 2D close-packed assembly of polystyrene (PS) nanospheres.66

Left panel: conceptual illustration showing plasmon gap-mode trapping with Au twin-pyramid nanostructure. Center panel: pictorial illustration of a 2D colloid crystal (green color) on plasmonic nanostructure (many small dots on the back); right panel: fluorescence image of polystyrene nanosphere (d=500 nm) assembly. Reprinted with permission from Shoji et al.66 (Copyright 2013, American Chemical Society).
As expected for optical trapping, the 2D colloidal crystal formation is reversible and is only formed during laser illumination. With the aid of a plasmonic structure (Figure 13), the laser intensity for stable trapping decreased to ~103 W cm−2, which compares favorably with that of conventional optical tweezers of ~107–108 W cm−2. Fluorescence microscopy imaging revealed that the far-field fluorescence intensity as a function of trapping laser intensity exhibited a maximum at 2.0 × 103 W cm−2 for fluorescent PS particles with diameters from 100 to 500 nm and decreased at higher intensities, suggesting an optimum trapping force exists to maintain a stable assembly (Figure 13, right). Moreover, along with the optical force that draws the particles together, photothermal effects are assumed to come into effect because of light absorption by the host Au NS. Of the photothermal effects, thermophoresis is supposed to act as a repulsive force opposing the radiation force. Furthermore, the force from thermal convection benefits trapping. The observation of a 2D crystal structure demonstrates that plasmon-enhanced optical trapping is a promising method for fabricating microscale crystals of NPs. Although the optical force increases with increasing laser power, the power should be kept at an optimum level because of the repulsive force that acts when the Au NS is heated significantly. In this example, the contribution from the photothermal effect is minor and can be controlled by adjusting the trapping laser power.
The Au NS characterized by twin-pyramid arrays (Figure 13, left) has an  (the square of the electric field) enhancement factor of 103−104, if light of a wavelength that coincides with the optical extinction maximum of ~800 nm is irradiated to form an optical trap.66 Other platforms such as Au nano-islands and random arrays of particles have been employed to gather/assemble particles. For instance, a close-packed Au NP film on a glass coverslip immersed in a solution containing PS beads or DNA was irradiated with a laser with a wavelength of 633 nm (< 25 mW) on a microscope stage.67 Convective-flow-induced concentration was demonstrated with PS beads with a diameter of 590 nm and λ DNA (8.4 kbp). Once the particles were concentrated, they remained stable at the location, likely due to surface adsorption. In this example, thermal convection was presumed to dominate in establishing the optical trapping. The interpretation contrasts that of the Tsuboi group.66 Using Au nano-island substrates, another study demonstrated the optical trapping and assembly of PS microspheres and living cells into highly organized patterns with low laser power density (~104 W cm−2 at 785 nm).68 The observed trapping is attributed to the net contribution from a near-field optical trapping force and a long-range thermophoretic force that overcomes the axial convective drag force. In this case, the thermophoretic force was assumed to have dragged the PS particles from cold to hot, the opposite direction to that suggested by other studies.58, 66 To realize plasmon-assisted optical trapping, hot spots that exhibit enormous enhancements are a prerequisite.55, 56 For Au nano-island films, reproducible formation of such hot spots may largely depend on technical ability. This can result in differing optimum trapping strengths depending on the sample.
(the square of the electric field) enhancement factor of 103−104, if light of a wavelength that coincides with the optical extinction maximum of ~800 nm is irradiated to form an optical trap.66 Other platforms such as Au nano-islands and random arrays of particles have been employed to gather/assemble particles. For instance, a close-packed Au NP film on a glass coverslip immersed in a solution containing PS beads or DNA was irradiated with a laser with a wavelength of 633 nm (< 25 mW) on a microscope stage.67 Convective-flow-induced concentration was demonstrated with PS beads with a diameter of 590 nm and λ DNA (8.4 kbp). Once the particles were concentrated, they remained stable at the location, likely due to surface adsorption. In this example, thermal convection was presumed to dominate in establishing the optical trapping. The interpretation contrasts that of the Tsuboi group.66 Using Au nano-island substrates, another study demonstrated the optical trapping and assembly of PS microspheres and living cells into highly organized patterns with low laser power density (~104 W cm−2 at 785 nm).68 The observed trapping is attributed to the net contribution from a near-field optical trapping force and a long-range thermophoretic force that overcomes the axial convective drag force. In this case, the thermophoretic force was assumed to have dragged the PS particles from cold to hot, the opposite direction to that suggested by other studies.58, 66 To realize plasmon-assisted optical trapping, hot spots that exhibit enormous enhancements are a prerequisite.55, 56 For Au nano-island films, reproducible formation of such hot spots may largely depend on technical ability. This can result in differing optimum trapping strengths depending on the sample.
Optical-trapping-based fabrication on plasmonic NSs was applied to prepare a ring structure. A DNA ring of approximately the size of a focused laser spot was found to form permanently on a focused CW-laser (λ=808 nm) illumination assisted by the Au twin-pyramid structure at an intensity of 5 × 103 W cm−2.69 Interestingly, for femtosecond laser irradiation (λ=770 nm, 80 MHz, 120 fs), only reversible trapping and release was observed. The mechanism behind this structural formation was not fully uncovered but the balance of an attractive gradient force and repulsive thermophoretic force because of accumulated heat may contribute to the structure. Phenomenologically, the ring formation is reminiscent of the microscale thermophoresis-based accumulation of DNA by the Braun group, as mentioned earlier (Scheme 2).58 In this context, it was reported that colloidal Au nanorods near the substrate surface can undergo photothermal trapping with a femtosecond laser at the LSPR wavelength (840 nm).70 Here, the trapping and immobilization of Au nanorods was demonstrated on a bare glass surface with 0.5 mW power. The mechanism is similar to Scheme 2, with convection and thermophoresis postulated as causing medium heating through heat transfer from the nanorods. By adjusting the distance between the laser focus and glass surfaces, immobilized ring patterns of Au nanorods were observed on the surface. These ring patterns could be controlled by using the trapping laser power and the numerical aperture of the objective. Although the observation of a ring pattern was very similar to that by the Tsuboi group,69 a contrasting mechanism that is entirely photothermal with no optical trapping force was postulated because of the absence of plasmonic substrates. However, we note that the tight focus with an NA 1.2 objective may sometimes generate optical gradients and scattering forces sufficient to form patterned aggregates on the substrates depending on the laser power.
In optical trapping, 3D trapping has been considered superior because of the greater degree of confinement.18 Nevertheless, in plasmon-enhanced optical trapping, substrates that support plasmonic structures were introduced, which imposed an optical trapping restricted to two dimensions. The substrates eventually formed an interface to regulate internal flow within the chamber as well as a site to accumulate molecules and colloids, resulting in meaningful structures for later use.71
Without plasmonic NSs, optical forces were used for nanofabrication by fully exploiting the plasmonic properties of the target NPs under intense laser illumination (~106 W cm−2), which may raise the NP temperature to near or greater than the melting point. Here, active use of bare substrates as platforms was made in pursuit of plasmon-assisted chemistry or material fabrication. When optical forces are applied to a colloidal Au NP solution to bring it close enough to a substrate, an attractive force between the substrate and the particle dominates, and as a result, the NP is fixed on the substrate. This optical trapping-based fabrication method is called laser/optical printing.72 Optical printing has been limited to the fabrication of ordered arrays of isolated NPs, that is, with a minimum interparticle distance of ~300 nm. A recent study proposed that the repulsive interaction that prevents the printing of NPs at close distance is produced by thermophoretic repulsion resulting from plasmonic heating of the already-printed NPs.73 The study developed a protocol to overcome this obstacle and fabricated connected NPs with well-defined orientation. To fabricate Au–Ag dimers (Figure 14, upper panels), the order of printing is the most important aspect.

Upper panels: schematic of the process for dimers fabrication (left): first Ag NPs were optically printed with the 405-nm laser. Then the colloidal suspension is replaced by Au NPs, which are subsequently printed using the 532-nm laser. Dark field (center) and FE-SEM (right) images of Ag−Au heterodimer. Reprinted with permission from Gargiulo et al.73 (Copyright 2016, American Chemical Society). Lower panels: Optical bending and printing of gold nanorods. (a) Schematic illustration of the bending process. Gold nanorods are diffusing in solution (step 1). A single rod aligns horizontally as soon as it enters the laser focus (step 2). The nanoparticle is heated in the laser beam, while optical forces simultaneously push the particle toward the substrate (step 3). (b) Bending angle of the printed particles can be controlled by the laser power density. Optically printed gold nanorods display a transition from a straight (*) to a bent (**) morphology above a laser power density of 0.45 × 106 W cm−2. The red dotted line indicates the laser power density that is required to heat the nanorods above their melting temperature according to simulations. (c) SEM images of gold nanorods that were printed with different bending angles. Scale bar is 100 nm. Reprinted with permission from Babynina et al.74 (Copyright 2016, American Chemical Society).
The repulsive force can be significantly reduced by printing Au NPs via excitation at 532 nm on already-printed Ag NPs that do not absorb the excitation light. Thus, the heating-induced repulsion was minimized during printing, resulting in connected NPs with well-defined orientation. This result suggests the importance of regulating mutual repulsive interactions of particles in respect to relatively weak optical forces.
As another example using a bare substrate for plasmonic NPs, it was demonstrated that Au nanorods can be bent in a controlled manner via focused laser illumination near the substrate surface (Figure 14, lower panels).74 A combination of optical forces with plasmonic heating rendered it possible to adjust the bending angle of single gold nanorods by changing the laser power.
Figure 14 (lower panel, a) presents a schematic of the experiment. Au nanorods (124 × 21 nm) were immersed in water and illuminated by a NIR laser beam (λ=1064 nm) that was focused a few microns above the substrate. The wavelength of the laser beam was set to the resonance wavelength of the longitudinal plasmon of the rods. Because the scattering force is strong, a single rod was pushed along the direction of the propagating light. Au nanorods were printed one by one on the substrate. By gradually increasing the laser power, the straight nanorods were bent (Figure 14, lower panel, b and c). Bent structures were observed for laser power densities greater than 4.5 × 105 W cm−2, which is sufficient to heat the particles above the melting point of gold. Bending was only observed for particles that were freely diffused in solution.
Through optical printing of colloidal NPs, the plasmon-assisted and polarization-directed growth of Au NPs from spheres to spheroids was achieved.75 In the first step, single Au NPs 60 nm in diameter were optically printed on a glass substrate from a colloidal suspension. The second step exchanged the Au NP colloid with an aqueous solution of HAuCl4 (1.5 μM). Finally, each NP was irradiated with linearly polarized light of 532 nm (9 × 105 W cm−2). The light-induced growth occurs preferentially in the direction of the electric field of the light used for the growth reaction, as confirmed both by dark-field scattering spectral peak shifts and the morphology of the Au NPs characterized by SEM. The mechanistic details of light-induced growth are non-trivial and are still a matter of investigation. Three possibilities are postulated: hot-electron-induced reduction of AuCl4−, plasmon-enhanced photoreduction of AuCl4− and plasmonic-heating-induced thermal reduction of AuCl4−. Temperature estimations suggest that during the growth reaction, the Au NP surface and the surrounding solution are at temperatures considerably higher (ca. 210 K) than room temperature. Nevertheless, Violi and coworkers assumed that the temperature increase does not play a dominant role in the growth reaction.75 Because the temperature of a NP is homogeneous over its surface, a thermally induced reaction would lead to isotropic and polarization-independent growth, which is in contrast to their observations.
A relevant study to that given above was reported,76 in which optical forces were used to shape particles without changing their volume. In this case, an optical force was used to elongate a single spherical Au NP into a rod through exposure to a focused Gaussian laser beam with a wavelength of 515 nm that excited the LSPR band (Figure 15).

Left: scattering spectral change under dark-field illumination of the melted gold nanoparticles from green to red. The corresponding SEM particle images from spherical to elongated particles are shown. Right: a model of the particle elongation induced by laser light. The Au NP in the center of the focused Gaussian beam is exposed to the radiation pressure force. The scattering force has the same direction as the laser beam pulling the particle upwardly. The gradient force arises from a strong gradient of the electric field and squeezes the particle perpendicular to the laser beam. Reprinted with permission from Kuhlicke et al.76 (Copyright 2013, American Chemical Society).
The laser-induced melting of gold in a tightly focused laser beam in combination with the radiation pressure of the focused light is considered to be responsible for the elongation. The optical properties and shape were measured via dark-field scattering spectra, SEM and AFM. Spherical Au NPs with a diameter of ~80 nm on a glass substrate in air began to melt at a laser intensity of 8 × 105 W cm−2 and were destroyed when the laser intensity exceeded 2.0 × 106 W cm−2. Hence, the experiment was performed with intermediate intensities.
In the above examples, optical forces are responsible for assembling and shaping NPs, but the contribution of the photothermal effect cannot be neglected. Presumably, synergy between the optical force and photothermal properties plays a role.
Plasmonic heating-based fabrication
Here we describe plasmonic fabrication, in which photothermal effects play a major role. Plasmon-enhanced thermophoresis allowed the light-directed reversible assembly of plasmonic nanotriangles (Figure 16a).77

(a) Schematic representation of light-directed reversible assembly of plasmonic nanotriangles based on plasmon-enhanced thermophoresis. (b) SEM image of Au nano-islands substrate used as a platform for the experiment. (c) Microscope images before laser illumination (upper), and after 10 s illumination (lower). Scale bar: 10 μm. (d) Schematic illustration of the migration of a CTA+-modified Au nanotriangle from cold to hot region in the light-induced temperature-gradient field, which is known as thermophoresis. (e) Schematic illustration of the release or redispersion of a Au nanotriangle assembly due to electrostatic repulsive interaction when the laser is off and the temperature-gradient field disappears. Reprinted with permission from Lin et al.77 (Copyright 2016, American Chemical Society). (f) Working principle of photothermal bubble manipulation to fabricate patterned colloidal particles (left). Dark-field optical images of the ‘BPL’ (960 nm PS beads, upper right) and ‘SP’ patterns (540 nm PS beads, middle right). Fluorescence lifetime image, ‘NANO’ from the patterned CdSe/ZnS dots (lower right). Reprinted with permission from Lin et al.78 (Copyright 2015, American Chemical Society).
To drive thermophoresis, a plasmonic Au nano-island substrate (Figure 16b) was optically heated through a microscope objective (NA: 0.85) forming a temperature-field gradient. As a result, surface-functionalized NPs were driven into the hot region on top of the substrate (Figure 16c, the upper frame (t=0 s) to the lower frame (t=10 s)). The utilized laser power density of ~3 × 103 W cm−2 (at λ=532 nm) is at least three orders of magnitude lower than that used in optical trapping for nanoassembly, and thus, thermophoretic trapping uses a lower optical power than optical trapping. According to Lin and coworkers,77 the driving mechanism was described as follows: in the presence of a temperature-field gradient, the asymmetric ion distribution in the electric double layer exerts a pressure and causes a slip flow of the liquid. Because of the positive DT (micelle) value, the CTAC micelles (CTAC concentration: 10 mM, cmc: 0.13 mM) move from the hot region to the cold region, while the liquid flow moves in the opposite direction, from the cold region to the hot region (Figure 16d). Here, the higher DT (micelle) value, compared with DT (Cl−), quickly drives the positive CTAC micelles toward the cold region and leads to the separation of negative and positive ions, thereby generating a temperature-dependent local electric potential. This exerts an electric force on the positive Au nanotriangles and drives the migration of the particles toward the hot region to form assemblies. When the laser is off and the temperature-gradient field disappears, the release or redispersion of the Au nanotriangle assembly occurs due to electrostatic repulsive interactions (Figure 16e). The reversible assembly of nanotriangles is seen because CTAC cannot dissociate from Au nanotriangles under illumination at a sufficient concentration. As an application of the method, parallel and dynamic manipulation of multiple NP assemblies was demonstrated using a holographic optical system.
In another study, optically controlled microbubbles were used to pattern colloidal particles onto plasmonic substrates.78 A single laser beam generates a microbubble at the interface of the colloidal suspension and the substrate (Au nano-island film) via plasmon-enhanced heating. The microbubble captures and immobilizes the colloidal particles on the substrate (Figure 16f, left) through the coordinated actions of Marangoni convection, surface tension, gas pressure and substrate adhesion. The study demonstrated that by directing the microbubble with the laser beam, one can create arbitrary single-particle patterns and particle assemblies with different architectures (Figure 16f, right).
Using a plasmonic NS acting as a photothermal heat source, an experimental technique was developed to perform the solvothermal synthesis of nano- and microcrystals in superheated water at ambient pressure.79 Solvothermal reactions occur normally in metastable liquids above their boiling point. Au nanorings on a glass substrate (Figure 17a) were illuminated with a laser beam 10 μm in diameter, reaching a maximum temperature close to 200 °C (473 K) (Figure 17b) and resulting in the synthesis of In(OH)3 microcrystals (Figure 17c).

(a) Gold nanorings on a glass substrate prepared by block copolymer micellar lithography. (b) Associated temperature distribution when the sample is illuminated with a laser beam (λ=800 nm), 10 μm in diameter, reaching a value close to 200 °C at the center of the heated area. (c) SEM image of In(OH)3 microcrystals formed upon heating an area of 10 μm in diameter (laser exposure time=1 h, laser power=18.5 mW, maximum temperature=200 °C, and reactant concentration=4 mM). Reprinted from Robert et al.79.
Compared with Au nano-islands that mainly absorb 550–600 nm light, the Au nanorings exhibit an absorption peak at ~800 nm. An advantage of plasmonic heating via Au nanorings is that water does not boil at 100°C (373 K) and vapor bubble formation systematically occurs at approximately 493±10 K, enabling solvothermal reactions under atmospheric pressure and temperatures up to 200 °C (473 K) without the use of an autoclave. This is the first demonstration of plasmonic heating-induced crystallization and opens possibilities to photothermally fabricate crystals using Au NSs without relying on autoclaves.
Instead of plasmonic substrates, such as Au NSs and Au nano-islands, optical heating-induced fabrication was pursued using single Au NPs supported on transparent substrates. The single-particle system is unique in that highly localized temperature gradients confined to nanoscale regions were formed around the NPs. Moreover, in terms of their varying thermal conductivities, the substrate plays a decisive role in controlling particle temperature, which is determined by radial heat conduction at the particle/medium and particle/substrate interfaces.80 For steady-state illumination, an example is given in Figure 18a for the 2D temperature distribution obtained by the numerical solution of the heat conduction equation. The radial temperature profile thus obtained was nearly concentric (Figure 18a, left) when the Au NP was supported on glass (kglass=1.0 W m−1 K−1) and exposed to water (kwater=0.60 W m−1 K−1). By contrast, when a sapphire substrate was used, a strongly distorted temperature distribution was expected because of the strong thermal conductivity of ksapphire=42 W m−1 K−1 (Figure 18a, right). Thus, to obtain a particle temperature equivalent to that for glass/water, a laser intensity approximately three times greater is needed for sapphire/water. Based on the numerical temperature simulations, the laser peak power density vs. particle temperature (Figure 18b) was linear with a gradient three times greater than that of the glass substrate, suggesting remarkable cooling in the sapphire substrate. Notably, because the profile is non-centrosymmetric, temperatures in the area below the particle are expected to be appreciably higher than those above the particle (Figure 18c). Consequently, a surface tension gradient can be generated because of the temperature gradient along the particle surface from the lower half of the particle in the direction of the substrate and the upper half of the particle. This surface tension gradient may be the origin of the slip flow along the particle surface to the substrate, known as the Marangoni convective flow (Figure 18d).59 Here, we note that the 2D temperature profiles in Figure 18a are typical for this scale in which a point-like heat source (~100 nm) is surrounded by an almost infinite-scale heat sink (cm × cm × ~100 μm). Because the high-temperature region is confined, thermal convection is predicted to be slow (Figure 18e).

(a) Simulated 2D temperature distributions for water-immersed 100-nm Au NP on glass (left side) and on sapphire (right side), under optical illumination from a CW laser (λ=488 nm). Up beam configuration was assumed to illuminate a Au NP supported on the top wall (ceiling) of the sample chamber. (b) Calculated particle temperature as a function of applied laser peak power density for water-immersed 100-nm-Au NP on sapphire (red line) and glass (black line) substrates. (c) Calculated 1D temperature profiles along upper (z=0) and lower (z=−100 nm) sides of the particle (100 nm in diameter), showing that a Au NP supported on sapphire substrate is cooler on the upper side (laser peak power density: 5.3 mW μm−2). (d) Schematic illustrating plasmonic-heating-induced Marangoni convective flow. (e) The 2D temperature distribution and the emerging free convective flow (white arrows) from optical heating of a 100-nm diameter Au NP (simulation). The particle temperature was set to 500 K that generated a flow velocity of 2.4 nm s−1 in front of the Au NP. Note that 1.0 mW μm−2=105 W cm−2=109 W m−2. Reprinted from Aibara et al.70 (f) Time-dependent LSPR red shifts with enhanced scattering spectral intensities for a single 100-nm-diameter Au NP at 4-s irradiation intervals for 5.0% PEG 6000 solution on a sapphire substrate. SEM images of (g) a bare Au NP and (h) a Au NP irradiated in the presence of 5.0% PEG. The shells were formed with 10 illumination cycles of 4 s (total 40 s) and a laser peak power density of 8.8 mW μm−2. Reprinted from Enders et al.81.
From the viewpoint of single-particle heating, we examine the recent observation of plasmonic-heating-induced fabrication by Enders et al.81 They used a 100-nm-diameter single Au NP supported on sapphire (2D temperature profile: Figure 18a, right) as a platform for the formation of NSs. They observed the transport of molecules such as polyethylene glycol (molecular weight of 6000 g mol−1, abbreviated PEG 6000) and sodium dodecyl sulfate (SDS) toward the Au NP, which then attached to its surface. They formed a gold core–organic shell structure within several tens of seconds after illumination by a 488-nm laser, which raised the particle temperature to ~360–400 K. Spectroscopically, because of the gradual increase in encapsulation, progressive redshifts with enhanced scattering intensities are seen in the LSPR bands of the Au NP with increasing illumination cycles (Figure 18f). Post-mortem SEM provided direct evidence of shell formation (compare the before and after images in Figures 18g and h). The observation of nanoscale heating-induced accumulation of polymers around the NP was unexpected due to the presumption that the experimental laser intensities of <106 W cm−2 were insufficient for the optical trapping of such molecules without plasmonic substrates. Alternatively, the optical-heating-induced Marangoni and thermal convections were considered likely to cause molecules to be carried toward the particle.
The observation that single Au NP heating induced the fabrication of core-shell structures prompted a study of thermoresponsive polymers to promote phase transition/phase separation for heating at the nanoscale.71 Such possibility was tested with poly(N-isopropylacrylamide) (PNIPAM), which is a well-investigated water-soluble thermoresponsive polymer.82 At room temperature, PNIPAM dissolves in water in a random coil state forming a homogeneous phase; when heated to temperatures above 32 °C (305 K)—the lower critical solution temperature (LCST)—a phase transition occurs from a coil to a globule state. The globular molecules aggregate due to hydrophobic interactions, resulting in phase separation and causing the solution to turn turbid. Previously, microscopy observations revealed that the phase transition of PNIPAM resulted in microparticle formation when aqueous solutions were heated directly with a focused infrared laser.83, 84 This was also observed in D2O, for which the absorption of infrared light is negligible; the PNIPAM formed microparticles in response to the optical force.83, 84
Figure 19a shows the single-particle Rayleigh scattering spectral changes originating from the heating of a Au NP in 1.0% aqueous PNIPAM solution. At a laser peak power density of 1.8 × 104 W cm−2 (0.18 mW μm−2), the spectral shift was within experimental error, whereas appreciable redshifts at 3.5 × 104 W cm−2 and 5.3 × 104 W cm−2 were recorded. Notably, these shifts occurred only during illumination; when stopped, the original spectra returned. For plasmon-assisted optical trapping, although not a heating effect, the reversible aggregation of PNIPAM on a Au NS substrate, forming microparticles, was observed microscopically at intensities as low as ~103 W cm−2.85

(a) In situ scattering spectral changes: before, during, and after laser illumination of a 100-nm-diameter Au NP supported on a glass substrate and submerged in aqueous solution of 1.0% PNIPAM. (b, c) Scattering spectra during the illumination of 488-nm laser light at laser peak power densities, 0.18, 0.35 and 0.53 mW μm−2, together with spectra before and after irradiation. Right panel: single-particle scattering spectrum with corresponding SEM image (inset) of (b) pristine Au NP and (c) subsequent changes arising from laser illumination after irradiation at 5.3 mW μm−2 for 8 s. Reprinted from Aibara et al.71.
The origin of the spectral shifts in the PNIPAM/Au NP system is ascribed to an increase in the refractive index sensed by the Au NP.86 The phenomenon was interpreted as the LCST-induced aggregation of PNIPAM around the NP due to heating of the surrounding medium by heat transfer from the optically heated NP. PNIPAM molecules are hydrophilic at temperatures below the LCST (305 K) but hydrophobic above the LCST.82 The phase transition and aggregation of PNIPAM may increase the refractive index of the medium sensed by the Au NP, resulting in LSPR redshifts. Remarkably, the reversible spectral changes observed at low laser intensities became irreversible when the laser intensity exceeded the specified limit (2.6 × 105 W cm−2 on a sapphire substrate). As a result, shell formation occurred around the Au NP, as evidenced in the SEM images, which is consistent with the permanent scattering spectral redshift (Figures 19b and c). Polymer-shell formation around the Au NP can be regarded as due to liquid–liquid phase separation and accumulation. This phenomenon is intriguing because accumulation occurs around the hot NP. The advantage of single plasmonic NP heating-induced fabrication is that in situ spectroscopic monitoring of the reaction is feasible before SEM analysis.
Single plasmonic NP heating-induced glass structuring was implemented by Osaka and coworkers.87 They demonstrated the fabrication of nanoholes on glass surfaces by embedding Au NPs under photoillumination. As illustrated in Figure 20 (left), single Au NPs supported on a borosilicate glass substrate submerged in aqueous alkaline solution, such as tetrabutylammonium hydroxide (TBAOH), were subjected to illumination with a focused laser beam that excites the LSPR band of the NPs. The SEM images (Figures 20a–f, center) exhibit the effect of laser exposure on the glass surfaces. Before illumination (Figure 20a), the particle is supported on the glass surface; after illumination for 10 s, the Au sphere sinks into the substrate, fitting just inside a cavity (Figure 20b). By increasing the irradiation period to 30 s (Figure 20c) or 30 min (Figure 20d), deeper holes are fabricated; the particle is seen in Figure 20c but not in Figure 20d. When a Au nanotriangle was used instead of a Au nanosphere, a nanohole with a triangular face was fabricated (Figure 20e). However, when illuminated at a lower laser intensity of 1.7 mW μm−2—one half of that used in this instance (Figure 20f)—the Au nanotriangle remains on the glass surface without forming a nanohole. This suggests that a threshold intensity exists for nanohole fabrication. On silica glass, nanoholes were fabricated in the same manner as for borosilicate glass.

Left: experimental outline for plasmonic-heating-induced nanohole fabrication on glass substrates. Center: SEM images showing the laser-induced embedding into glass substrate for illumination with a peak power density of 5.0 mW μm−2 on 100-nm-diameter Au nanospheres (a–d) and Au nanotriangles (e, f). (a) before irradiation, Au NP is sitting on the substrate surface; (b) after 10 s of irradiation, Au NP is fallen into a hole; (c), after 30 s of irradiation, Au NP is seen at the bottom; (d), after 30 min of irradiation, Au NP is far inside the hole and could not be seen. (e) after irradiation at 180 s at 3.3 mW μm−2, Au nanotriangle is embedded; (f) after irradiation for 180 s at 1.7 mW μm−2, no embedding of Au nanotriangle is seen. All scale bars represent 100 nm. Right: dark-field optical microscope images (upper panels) and corresponding cross-sectional schematics (lower panels) of the steps used to create a horizontal nanohole by manipulating a Au NP using the laser focus spot. First, a Au NP, orange spot at the center of (g) supported on a glass substrate was embedded with focused laser illumination of intensity 3.3 mW μm−2 for 40 min. After embedding, the laser focusing spot was moved horizontally in small steps; after 120 min (h), we see the Au NP is 2 μm from the nanohole entrance (circled). The scale bars represent 2 μm. Reprinted with permission from Osaka et al.87 (Copyright 2016, The Royal Society of Chemistry).
The susceptibility to etching of the glass surface is ascribed to laser-heating-induced structural modification because heat transfer from the particle causes the temperature of the glass surface to increase. The steady-state particle temperatures reached were estimated to be 400−500 K. During the alkali etching of glass, [OH−] plays a crucial role for facile processing. In this regard, the ion product of water, Kw is temperature-dependent. As the temperature increases from 298 K to 400 and 500 K, Kw increases from 10−14.0 to 10−11.9 and 10−11.2.79 The resulting higher [OH−] should markedly increase the etching rate of glass.
To embed the particle forcefully and fabricate nanoholes in desired directions, an optical manipulation technique was applied (Figures 20g and h). First, a Au NP (orange spot at the center) supported on a glass substrate (Figure 20a) was illuminated, and the particle was embedded. Then, the focal spot of the laser beam (d=~1 μm) was moved near the embedded particle to shift it laterally in the direction the focal spot is moving, to a distance ~2 μm from the entrance after 120 min of illumination (Figure 20b). Presumably, the scattering force from the laser beam pushes the particle.88 Using diffraction-limited optics and a piezo stage would improve this form of manipulation.
Aside from this etching-assisted nanohole fabrication using an embedded Au NP, mere homogeneous heating in an oven embeds Au NPs into SiO2 glass. Au particles with an initial diameter of 1 μm and thickness of 18 nm were buried into SiO2 glass when heated to 1050 °C, leaving nanoholes, as reported by de Vreede et al.89 After 9 hours of heating, a 0.8-μm-deep nanohole was formed, reducing the Au NP diameter to 25 nm. Evaporation of the Au NP occurs during the process as the temperature was close to the melting point of Au (1064 °C). The driving force in the embedding is the continual evaporation of Au transporting SiO2 to the surface, which led to ridge formation. In practice, their method is hindered by its slowness, unfavorable ridge deposits and the channel narrowing with depth. For nanohole fabrication, etching is free from these shortcomings. Interestingly, Hidai and coworkers90 used a system similar to that of de Vreede and coworkers but instead illuminated the glass substrate with a CW laser (λ=514 nm) horizontally from the opposite side, such that the migration of metal microparticles occurs but no holes are produced. In this instance, a liquid droplet of the metal moves within the glass toward the laser direction, softening the glass ahead of the moving particle. The void created behind the particle is filled by the softened glass and rapidly cooled at ambient temperature. The result suggests that etching reagents are essential for glass structuring. Furthermore, plasmonic heating combined with etching reagents is useful for triggering and terminating fabrication as well as controlling the etching rate using light.
Plasmonic field enhancement-based fabrication
Energy-saving plasmonic enhancement has been applied for two-photon and multi-photon-mediated photochemistry:62 photoisomerization, photopolymerization and photomodification. Here we give examples of unconventional structuring achieved through multi-photon processes.
On DNA nanowires prepared on a poly(methyl methacrylate) (PMMA) film, plasmon-mediated excitation transfer over one micron was demonstrated with Au NPs attached to a double-stranded DNA (dsDNA) excited using a femtosecond laser pulse.91 AFM images (Figure 21, upper left frames) show the effects of the femtosecond laser pulses at λ=1030 nm, clearly indicating that only the parallel alignment of the excitation electric field is effecting long-range changes. Upon illumination using diagonally polarized laser pulses, only local damage was seen (Figures 21a and b). By contrast, extensive damage was observed in the DNA strand when illuminated by vertical/horizontally polarized pulses (Figures 21c and d). Underlying this observation is the plasmonic excitation-induced escape of electrons, which damages the DNA strand and the PMMA support as well as embeds the Au NPs into the film. These plasmonic excitation-induced events observed using femtosecond lasers were categorized into three groups: (1) LSPR excitation; (2) single Au NP exaction-induced electron emission into the host PMMA and damage; and (3) excitation transfer from Au NP multimers (dimers, trimers) to DNA, resulting in extensive damage (Figure 21, upper right frames). In explaining the observed ablation, photothermal effects were eliminated based on temperature calculations giving a rise of only 40 K at most, as this temperature is too low to embed a metal NP in a PMMA layer compared with the required temperature of 300 K. Their main point is that the plasmon-enhanced strong electric field can drive NP electron tunneling based on four-photon excitation using a laser wavelength of 1030 nm (1.20 eV) to overcome the work function of gold (~5.5 eV).

Upper left: AFM images showing the effects of electric field alignment (with respect to the long axis of nanoparticle multimers) on excitation transfer along DNA (height scale: 15 nm). For diagonal polarization, only local damage along the DNA is observed (~200-nm fragmentation); only the particle is present before irradiation (a) and disappears into the substrate (b). When the field is aligned (horizontal polarization) (c), irradiation results also in extensive damage (~2-μm fragmentation) along the DNA (d). Upper right: Schematics showing three steps of excitation during femtosecond laser irradiation of metallic nanoparticles associated with surface phenomena and their effects on surroundings. Reprinted with permission from Wirth et al.91 (Copyright 2014, American Chemical Society). Lower frame: Polarized femtosecond laser(λ=800 nm, 120 fs)-mediated growth and programmable assembly of photoreduced silver nanoparticles into triply hierarchical micropatterns (thickness ~30 nm): (i) the excited surface plasmons enhance the local electric field and lead to spatially selective growth of silver atoms at the opposite ends of dipoles induced on early created silver seeds; (ii, iii) the optical attractive force overcomes electrostatic repulsion in the enhanced local electric field to assemble the silver nanoparticles directly. Reprinted with permission from Xu et al.92 (Copyright 2014, American Chemical Society).
Another example is the multi-photon-induced optical fabrication of hierarchically integrated metal NSs employing silver. In this research, the femtosecond-laser mediated growth and assembly of photoreduced Ag NPs into nanoplates and micropatterns were revealed (Figure 21, lower frame).92 First, the photoreduction of silver ions in solution in the presence of ammonia and trisodium citrate occurs through the multi-photon absorption of a 800-nm laser to form Ag NPs. Then, the LSPR fields were excited and established on the Ag seeds (far left), launching directional growth along the dipole ends by attracting atoms that were photoreduced in solution (i). Finally, nanoplates formed gradually (ii and iii), resulting in an arrayed structure (far right).
Thus, the production of nanoplate arrays was attributed to the local field enhancement induced by the LSPR mode of the Ag particles. One can eliminate the potential involvement of the ripple mechanism,93 that is, the periodic structures created by light interference, either between incident laser beams or between the incident beam and its excited waves. In the ripple formation, the patterns replicate the light intensity redistributions, and the structure periods depend on the laser wavelength and the beam incident angles.
Conclusion and future outlook
To date, the power conversion efficiency of plasmonic solar cells is still less than 1%, which is much lower than that achieved by current commercial solar cells (~20%), such as crystalline silicon cells. Plasmonic solar cells, however, have many attractive features, given that they can operate as a very thin film because of their very strong and widely tunable photoabsorption in the visible and NIR wavelength regions. Ultrafast spectroscopy has revealed that hot-electron transfer from plasmon-excited Au to a contacting semiconductor occurs within 100 fs, and the efficiency is between 20 and 80%. Hot spots generated through electric-field enhancements at specific positions in an Au NS potentially play an important role in hot-electron generation. If hot electrons approach the electron-accepting semiconductor, the transfer is efficient even in the presence of an intervening insulator barrier. Such an insulator layer or Schottky barrier in a semiconductor can suppress the unfavorable back electron transfer, increasing the photovoltage. Precise control over gold-interface semiconductor NSs would significantly increase the performance of future plasmonic solar cells.
Plasmonic photocatalysts will also continue to be an attractive research target because of their unique properties. Gold NSs on TiO2 are well recognized to be good photoanodes to produce O2 through the water-splitting reaction. H2 can also be produced in the presence of a Pt cocatalyst on TiO2. Their combination would enable a photocatalytic electrochemical sheet to split water into H2 and O2. As gold has a strong absorption, even small amounts can induce a sufficient visible-to-NIR response in conventional TiO2 catalysts to be used for environmental purification in hospitals or public buildings.94 To improve the efficiency of plasmonic solar cells and photocatalysts, it is very important to understand the primary processes in the reactions. With powerful femtosecond time-resolved and nanometer space-resolved spectroscopic techniques, further advancements may accelerate the development of plasmonic devices and materials. Ultrasmall photodetectors, optical switches for information technology95 and photocatalysts for cancer therapy96 would then emerge as new applications.
With optically driven nanofabrication taking advantage of plasmonic NPs and NSs, two approaches are being developed to avoid mixing photothermal effects and optical manipulation. To pursue pure optical trapping free from photothermal effects, Au NSs were fabricated on a Au film, which acted as an effective heat sink because of its large thermal conductivity (314 W m−1 K−1) while reflecting most of the incident laser light.97 Further improvements in suppressing heating can be achieved by using a film or substrate made of diamond, which has a thermal conductivity of 1000 W m−1 K−1. Another way of quenching thermal convection resulting from medium water heating is to maintain the temperature at ~4°C; with a vanishingly small thermal expansion coefficient, buoyancy forces are thereby suppressed.98 If thermal convection is minimized in plasmon-assisted trapping, the area of trapping is significantly reduced, leading to assemblies of much smaller scale. Photothermal trapping to form assemblies similar to those prepared by optical forces is unexpected but intriguing for less refined protocols. Still, the details of the driving mechanism are poorly understood. In particular, the details of thermophoresis, such as the direction of thermodiffusion, is not well understood.99 Further theoretical treatment of photothermal trapping is needed.
Nevertheless, plasmonic heating has found useful applications. For instance, single-particle heating can be effective for metal core-polymer shell fabrication and glass structuring. In addition, plasmonic heating-induced crystallization on Au NP assemblies is promising for the hydrothermal synthesis of various nanocrystals at atmospheric pressure. These new methods should be pursued with further sophistication. Nanofabrication techniques that fully exploit the plasmonic properties are advancing the field of optical manipulation-based materials science and technology, and we expect to see steady development over the next few years.

Concept of plasmon-enhanced optical trapping vs. conventional optical trapping. (Left) Optical trapping of a NP at the focal point of the laser beam. (Center) Scattering force that pushes the particle against the wall. (Right) plasmonic optical tweezers that can trap at the plasmonic active nanospace with significantly reduced laser powers.

Concept of thermophoretic trapping from central heating originating from an interplay of lateral thermophoresis (1), (4) and axial thermophoresis (3) with convection (2). Reprinted with permission from Braun and Libchaber58 (Copyright 2002, American Institute of Physics).
References
Link, S. & El-Sayed, M. A. Spectral properties and relaxation dynamics of surface plasmon electronic oscillations in gold and silver nanodots and nanorods. J. Phys. Chem. B 103, 8410–8426 (1999).
Link, S. & El-Sayed, M. A. Shape and size dependence of radiative, non-radiative and photothermal properties of gold nanocrystals. Int. Rev. Phys. Chem. 19, 409–453 (2000).
Hartland, G. V. Optical studies of dynamics in noble metal nanostructures. Chem. Rev. 111, 3858–3887 (2011).
Katoh, R., Furube, A., Yoshihara, T., Hara, K., Fujihashi, G., Takano, S., Murata, S., Arakawa, H. & Tachiya, M. Efficiencies of electron injection from excited N3 dye into nanocrystalline semiconductor (ZrO2, TiO2, ZnO, Nb2O5, SnO2, In2O3 films. J. Phys. Chem. B 108, 4818–4822 (2004).
Zhang, S., Yang, X., Numata, Y. & Han, L. Highly efficient dye-sensitized solar cells: progress and future challenges. Energy Environ. Sci. 6, 1443–1464 (2013).
Furube, A., Katoh, R. & Hara, K. Electron injection dynamics in dye-sensitized semiconductor nanocrystalline films. Surf. Sci. Rep. 69, 389–441 (2014).
Katoh, R. & Furube, A. Electron injection efficiency in dye-sensitized solar cells. J. Photochem. Photobiol. C 20, 1–16 (2014).
Tachibana, Y., Moser, J. E., Grätzel, M., Klug, D. R. & Durrant, J. R. Subpicosecond interfacial charge separation in dye-sensitized nanocrystalline titanium dioxide films. J. Phys. Chem. 100, 20056–20062 (1996).
Kale, M. J. & Christopher, P. Plasmons at the interface. Science 349, 587–588 (2015).
Wu, K., Chen, J., McBride, J. & Lian, T. Efficient hot-electron transfer by a plasmon-induced interfacial charge-transfer transition. Science 349, 632–635 (2015).
Foerster, B., Joplin, A., Kaefer, K., Celiksoy, S., Link, S. & Sönnichsen,, C. Chemical interface damping depends on electrons reaching the surface. ACS Nano 11, 2886–2893 (2017).
Tian, Y. & Tatsuma, T. Plasmon-induced photoelectrochemistry at metal nanoparticles supported on nanoporous TiO2 . Chem. Commun. 1810–1811 (2004).
Tian, Y. & Tatsuma, T. Mechanisms and applications of plasmon-induced charge separation at TiO2 films loaded with gold nanoparticles. J. Am. Chem. Soc. 127, 7632–7637 (2005).
Linic, S., Christopher, P. & Ingram, D. B. Plasmonic-metal nanostructures for efficient conversion of solar to chemical energy. Nat. Mater. 10, 911–921 (2011).
Clavero, C. Plasmon-induced hot-electron generation at nanoparticle/metal-oxide interfaces for photovoltaic and photocatalytic devices. Nat. Photon. 8, 95–103 (2014).
Brongersma, M. L., Halas, N. J. & Nordlander, P. Plasmon-induced hot carrier science and technology. Nat. Nanotechnol. 10, 25–34 (2015).
Panayotov, D. A. & Morris, J. R. Surface chemistry of Au/TiO2: Thermally and photolytically activated reactions. Surf. Sci. Rep. 71, 77–271 (2016).
Ashkin, A., Dziedzic, J., Bjorkholm, J. & Chu, S. Observation of a single-beam gradient force optical trap for dielectric particles. Opt. Lett. 11, 288–290 (1986).
Grier, D. G. A revolution in optical manipulation. Nature 424, 810–816 (2003).
Grigorenko, A., Roberts, N., Dickinson, M. & Zhang, Y. Nanometric optical tweezers based on nanostructured substrates. Nat. Photon.Nat. Photon. 2, 365–370 (2008).
Righini, M., Volpe, G., Girard, C., Petrov, D. & Quidant, R. Surface plasmon optical tweezers: tunable optical manipulation in the femtonewton range. Phys. Rev. Lett. 100, 186804 (2008).
Urban, A. S., Carretero-Palacios, S., Lutich, A. A., Lohmüller, T., Feldmann, J. & Jäckel, F. Optical trapping and manipulation of plasmonic nanoparticles: fundamentals, applications, and perspectives. Nanoscale 6, 4458–4474 (2014).
Mataga, N., Okada, T. & Masuhara, H. (eds) Dynamics and mechanisms of photoinduced electron transfer and related phenomena. (Elsevier B.V., Amsterdam, Netherland, 1992).
Grätzel, M. Photoelectrochemical cells. Nature 414, 338–344 (2001).
Nishijima, Y., Ueno, K., Yokota, Y., Murakoshi, K. & Misawa, H. Plasmon-assisted photocurrent generation from visible to near-infrared wavelength using a Au-nanorods/TiO2 electrode. J. Phys. Chem. Lett. 1, 2031–2036 (2010).
Nishijima, Y., Ueno, K., Kotake, Y., Murakoshi, K., Inoue, H. & Misawa, H. Near-infrared plasmon-assisted water oxidation. J. Phys. Chem. Lett. 3, 1248–1252 (2012).
Garcia de Arquer, F. P., Mihi, A., Kufer, D. & Konstantatos, G. Photoelectric energy conversion of plasmon-generated hot carriers in metal–insulator–semiconductor structures. ACS Nano 7, 3581–3588 (2013).
Mubeen, S., Lee, J., Lee, W., Singh, N., Stucky, G. D. & Moskovits, M. On the plasmonic photovoltaic. ACS Nano 8, 6066–6073 (2014).
Nakamura, K., Oshikiri, T., Ueno, K., Wang, Y., Kamata, Y., Kotake, Y. & Misawa, H. Properties of plasmon-induced photoelectric conversion on a TiO2/NiO p–n junction with Au nanoparticles. J. Phys. Chem. Lett. 7, 1004–1009 (2016).
Lee, W., Navarrete, J., Evanko, B., Stucky, G. D., Mubeen, S. & Moskovits, M. A plasmonic liquid junction photovoltaic cell with greatly improved power conversion efficiency. Chem. Commun. 52, 13460–13462 (2016).
Kojima, A., Teshima, K., Shirai, Y. & Miyasaka, T. Organometal halide perovskites as visible-light sensitizers for photovoltaic cells. J. Am. Chem. Soc. 131, 6050–6051 (2009).
Mali, S. S., Shim, C. S., Kim, H., Patil, P. S. & Hong, C. K. In situ processed gold nanoparticle-embedded TiO2 nanofibers enabling plasmonic perovskite solar cells to exceed 14% conversion efficiency. Nanoscale 8, 2664–2677 (2016).
Fujishima, A. & Honda, K. Electrochemical photolysis of water at a semiconductor electrode. Nature 238, 37–38 (1972).
Hisatomi, T., Kubota, J. & Domen, K. Recent advances in semiconductors for photocatalytic and photoelectrochemical water splitting. Chem. Soc. Rev. 43, 7520–7535 (2014).
Ueno, K. & Misawa, H. Plasmon-enhanced photocurrent generation and water oxidation from visible to near-infrared wavelengths. Asia Mater. 5, e61 (2013).
Ueno, K., Oshikiri, T. & Misawa, H. Plasmon‐induced water splitting using metallic‐nanoparticle‐loaded photocatalysts and photoelectrodes. ChemPhysChem 17, 199–215 (2016).
DuChene, J. S., Sweeny, B. C., Johnston‐Peck, A. C., Su, D., Stach, E. A. & Wei, W.D Prolonged hot electron dynamics in plasmonic‐metal/semiconductor heterostructures with implications for solar photocatalysis. Angew. Chem. Int. Ed. 53, 7887–7891 (2014).
Kodiyath, R., Manikandan, M., Liu, L., Ramesh, G. V., Koyasu, S., Miyauchi, M., Sakuma, Y., Tanabe, T., Gunji, T. & Dao, T. D. Visible-light photodecomposition of acetaldehyde by TiO2-coated gold nanocages: plasmon-mediated hot electron transport via defect states. Chem. Commun. 50, 15553–15556 (2014).
Ide, Y., Nagao, K., Saito, K., Komaguchi, K., Fuji, R., Kogure, A., Sugahara, Y., Bando, Y. & Golberg, D. h-BN nanosheets as simple and effective additives to largely enhance the activity of Au/TiO2 plasmonic photocatalysts. Phys. Chem. Chem. Phys. 18, 79–83 (2015).
Furube, A., Du, L., Hara, K., Katoh, R. & Tachiya, M. Ultrafast plasmon-induced electron transfer from gold nanodots into TiO2 nanoparticles. J. Am. Chem. Soc. 129, 14852–14853 (2007).
Du, L., Furube, A., Hara, K., Katoh, R. & Tachiya, M. Ultrafast plasmon induced electron injection mechanism in gold–TiO2 nanoparticle system. J. Photochem. Photobiol. C 15, 21–30 (2013).
Du, L. C., Furube, A., Yamamoto, K., Hara, K., Katoh, R. & Tachiya, M. Plasmon-induced charge separation and recombination dynamics in Gold-TiO2 nanoparticle systems: dependence on TiO2 particle size. J. Phys. Chem. C 113, 6454–6462 (2009).
Cushing, S. K., Li, J., Meng, F., Senty, T. R., Suri, S., Zhi, M., Li, M., Bristow, A. D. & Wu, N. Photocatalytic activity enhanced by plasmonic resonant energy transfer from metal to semiconductor. J. Am. Chem. Soc. 134, 15033–15041 (2012).
Tang, Y., Jiang, Z., Xing, G., Li, A., Kanhere, P. D., Zhang, Y., Sum, T. C., Li, S., Chen, X. & Dong, Z. Efficient Ag@ AgCl cubic cage photocatalysts profit from ultrafast plasmon‐induced electron transfer processes. Adv. Funct. Mater. 23, 2932–2940 (2013).
Bian, Z., Tachikawa, T., Zhang, P., Fujitsuka, M. & Majima, T. Au/TiO2 superstructure-based plasmonic photocatalysts exhibiting efficient charge separation and unprecedented activity. J. Am. Chem. Soc. 136, 458–465 (2013).
Wu, K., Rodríguez-Córdoba, W. E., Yang, Y. & Lian, T Plasmon-induced hot electron transfer from the Au tip to CdS rod in CdS-Au nanoheterostructures. Nano Lett. 13, 5255–5263 (2013).
Harutyunyan, H., Martinson, A. B., Rosenmann, D., Khorashad, L. K., Besteiro, L. V., Govorov, A. O. & Wiederrecht, G. P. Anomalous ultrafast dynamics of hot plasmonic electrons in nanostructures with hot spots. Nat. Nanotechnol. 10, 770–774 (2015).
Kumar, D., Lee, A., Lee, T., Lim, M. & Lim, D.-K. Ultrafast and efficient transport of hot plasmonic electrons by graphene for Pt free, highly efficient visible-light responsive photocatalyst. Nano Lett. 16, 1760–1767 (2016).
Zeng, P., Cadusch, J., Chakraborty, D., Smith, T. A., Roberts, A., Sader, J. E., Davis, T. J. & Gómez, D. E. Photoinduced electron transfer in the strong coupling regime: waveguide–plasmon polaritons. Nano Lett. 16, 2651–2656 (2016).
Kubo, A., Onda, K., Petek, H., Sun, Z., Jung, Y. S. & Kim, H. K. Femtosecond imaging of surface plasmon dynamics in a nanostructured silver film. Nano Lett. 5, 1123–1127 (2005).
Sun, Q., Ueno, K., Yu, H., Kubo, A., Matsuo, Y. & Misawa, H. Direct imaging of the near field and dynamics of surface plasmon resonance on gold nanostructures using photoemission electron microscopy. Light: Sci. Appl. 2, e118 (2013).
Sun, Q., Yu, H., Ueno, K., Kubo, A., Matsuo, Y. & Misawa, H. Dissecting the few-femtosecond dephasing time of dipole and quadrupole modes in gold nanoparticles using polarized photoemission electron microscopy. ACS Nano 10, 3835–3842 (2016).
Lehmuskero, A., Johansson, P., Rubinsztein-Dunlop, H., Tong, L. & Kall, M. Laser trapping of colloidal metal nanoparticles. ACS Nano 9, 3453–3469 (2015).
Spesyvtseva, S.E.S. & Dholakia, K. Trapping in a material world. ACS Photon. 3, 719–736 (2016).
Juan, M. L., Righini, M. & Quidant, R. Plasmon nano-optical tweezers. Nat. Photon. 5, 349–356 (2011).
Shoji, T. & Tsuboi, Y. Plasmonic optical tweezers toward molecular manipulation: tailoring plasmonic nanostructure, light source, and resonant trapping. J. Phys. Chem. Lett. 5, 2957–2967 (2014).
Qin, Z. & Bischof, J. C. Thermophysical and biological responses of gold nanoparticle laser heating. Chem. Soc. Rev. 41, 1191–1217 (2012).
Braun, D. & Libchaber, A. Trapping of DNA by thermophoretic depletion and convection. Phys. Rev. Lett. 89, 188103 (2002).
Louchev, O. A., Juodkazis, S., Murazawa, N., Wada, S. & Misawa, H. Coupled laser molecular trapping, cluster assembly, and deposition fed by laser-induced Marangoni convection. Opt. Express 16, 5673–5680 (2008).
Hou, L., Yorulmaz, M., Verhart, N. R. & Orrit, M. Explosive formation and dynamics of vapor nanobubbles around a continuously heated gold nanosphere. New J. Phys. 17, 013050 (2015).
Zheng, Y., Liu, H., Wang, Y., Zhu, C., Wang, S., Cao, J. & Zhu, S. Accumulating microparticles and direct-writing micropatterns using a continuous-wave laser-induced vapor bubble. Lab. Chip 11, 3816–3820 (2011).
Ueno, K. & Misawa, H. Surface plasmon-enhanced photochemical reactions. J. Photochem. Photobiol. C 15, 31–52 (2013).
Ueno, K. & Misawa, H. Fabrication of nanoengineered metallic structures and their application to nonlinear photochemical reactions. Bull. Chem. Soc. Jpn. 85, 843–853 (2012).
Haynes, C. L. & Van Duyne, R. P. Nanosphere lithography: a versatile nanofabrication tool for studies of size-dependent nanoparticle optics. J. Phys. Chem. B 105, 5599–5611 (2001).
Karakouz, T., Maoz, B. M., Lando, G., Vaskevich, A. & Rubinstein, I. Stabilization of gold nanoparticle films on glass by thermal embedding. ACS Appl. Mater. Interfaces 3, 978–987 (2011).
Shoji, T., Shibata, M., Kitamura, N., Nagasawa, F., Takase, M., Murakoshi, K., Nobuhiro, A., Mizumoto, Y., Ishihara, H. & Tsuboi, Y. Reversible photoinduced formation and manipulation of a two-dimensional closely packed assembly of polystyrene nanospheres on a metallic nanostructure. J. Phys. Chem. C 117, 2500–2506 (2013).
Miao, X., Wilson, B. K., Pun, S. H. & Lin, L. Y. Optical manipulation of micron/submicron sized particles and biomolecules through plasmonics. Opt. Express 16, 13517–13525 (2008).
Kang, Z., Chen, J., Wu, S.-Y., Chen, K., Kong, S.-K., Yong, K.-T. & Ho, H.-P. Trapping and assembling of particles and live cells on large-scale random gold nano-island substrates. Sci. Rep. 5, 9978 (2015).
Shoji, T., Saitoh, J., Kitamura, N., Nagasawa, F., Murakoshi, K., Yamauchi, H., Ito, S., Miyasaka, H., Ishihara, H. & Tsuboi, Y. Permanent fixing or reversible trapping and release of DNA micropatterns on a gold nanostructure using continuous-wave or femtosecond-pulsed near-infrared laser light. J. Am. Chem. Soc. 135, 6643–6648 (2013).
Chen, H., Gratton, E. & Digman, M. A. Self-assisted optothermal trapping of gold nanorods under two-photon excitation. Methods Appl. Fluoresc. 4, 035003 (2016).
Aibara, I., Mukai, S. & Hashimoto, S. Plasmonic-heating-induced nanoscale phase separation of free poly (N-isopropylacrylamide) molecules. J. Phys. Chem. C 120, 17745–17752 (2016).
Urban, A. S., Lutich, A. A., Stefani, F. D. & Feldmann, J. Laser printing single gold nanoparticles. Nano Lett. 10, 4794–4798 (2010).
Gargiulo, J.n., Cerrota, S., Cortés, E., Violi, I. L. & Stefani, F.D Connecting metallic nanoparticles by optical printing. Nano Lett. 16, 1224–1229 (2016).
Babynina, A., Fedoruk, M., Kühler, P., Meledin, A, Döblinger, M & Lohmüller, T Bending gold nanorods with Light. Nano Lett. 16, 6485–6490 (2016).
Violi, I. L., Gargiulo, J.n., von Bilderling, C., Cortés, E. & Stefani, F. D. Light-induced polarization-directed growth of optically printed gold nanoparticles. Nano Lett. 16, 6529–6533 (2016).
Kuhlicke, A., Schietinger, S., Matyssek, C., Busch, K. & Benson, O. In situ observation of plasmon tuning in a single gold nanoparticle during controlled melting. Nano Lett. 13, 2041–2046 (2013).
Lin, L., Peng, X., Wang, M., Scarabelli, L., Mao, Z., Liz-Marzán, L. M., Becker, M. F. & Zheng, Y. Light-directed reversible assembly of plasmonic nanoparticles using plasmon-enhanced thermophoresis. ACS Nano 10, 9659–9668 (2016).
Lin, L., Peng, X., Mao, Z., Li, W., Yogeesh, M. N., Rajeeva, B. B., Perillo, E. P., Dunn, A. K., Akinwande, D. & Zheng, Y. Bubble-pen lithography. Nano Lett. 16, 701–708 (2015).
Robert, H. M., Kundrat, F., Bermuúdez-Ureña, E., Rigneault, H., Monneret, S., Quidant, R., Polleux, J. & Baffou, G. Light-assisted solvothermal chemistry using plasmonic nanoparticles. ACS Omega 1, 2–28 (2016).
Setoura, K., Okada, Y., Werner, D. & Hashimoto, S. Observation of nanoscale cooling effects by substrates and the surrounding media for single gold nanoparticles under CW-laser illumination. ACS Nano 7, 7874–7885 (2013).
Enders, M., Mukai, S., Uwada, T. & Hashimoto, S. Plasmonic nanofabrication through optical heating. J. Phys. Chem. C 120, 6723–6732 (2016).
Halperin, A., Kröger, M. & Winnik, F. M. Poly (N‐isopropylacrylamide) phase diagrams: fifty years of research. Angew. Chem., Int. Ed. 54, 15342–15367 (2015).
Hofkens, J., Hotta, J., Sasaki, K., Masuhara, H. & Iwai, K. Molecular assembling by the radiation pressure of a focused laser beam: poly (N-isopropylacrylamide) in aqueous solution. Langmuir 13, 414–419 (1997).
Tsuboi, Y., Nishino, M., Sasaki, T. & Kitamura, N. Poly (N-isopropylacrylamide) microparticles produced by radiation pressure of a focused laser beam: a structural analysis by confocal Raman microspectroscopy combined with a laser-trapping technique. J. Phys. Chem. B 109, 7033–7039 (2005).
Shoji, T. & Tsuboi, Y. Plasmonic optical trapping of soft nanomaterials such as polymer chains and DNA: micro-patterning formation. Opt. Rev. 22, 137–142 (2015).
Mayer, K. M. & Hafner, J. H. Localized surface plasmon resonance sensors. Chem. Rev. 111, 3828–3857 (2011).
Osaka, Y., Sugano, S. & Hashimoto, S. Plasmonic-heating-induced nanofabrication on glass substrates. Nanoscale 8, 18187–18196 (2016).
Fedoruk, M., Lutich, A. A. & Feldmann, J. Subdiffraction-limited milling by an optically driven single gold nanoparticle. ACS Nano 5, 7377–7382 (2011).
de Vreede, L. J., van den Berg, A. & Eijkel, J. C. Nanopore fabrication by heating Au particles on ceramic substrates. Nano Lett. 15, 727–731 (2015).
Hidai, H., Matsushita, M., Matsusaka, S., Chiba, A. & Morita, N. Moving force of metal particle migration induced by laser irradiation in borosilicate glass. Opt. Express 21, 18955–18962 (2013).
Wirth, J., Garwe, F., Meyer, R., Csáki, A., Stranik, O & Fritzsche, W Plasmonically enhanced electron escape from gold nanoparticles and their polarization-dependent excitation transfer along DNA nanowires. Nano Lett. 14, 3809–3816 (2014).
Xu, B.-B., Wang, L., Ma, Z.-C., Zhang, R., Chen, Q.-D., Lv, C., Han, B., Xiao, X.-Z., Zhang, X.-L., Zhang, Y.-L., Ueno, K., Misawa, H. & Sun, H.-B. Surface-plasmon-mediated programmable optical nanofabrication of an oriented silver nanoplate. ACS Nano 8, 6682–6692 (2014).
Rebollar, E., de Aldana, J.R.V., Martín-Fabiani, I., Hernández, M., Rueda, D. R., Ezquerra, T. A., Domingo, C., Moreno, P. & Castillejo, M Assessment of femtosecond laser induced periodic surface structures on polymer films. Phys. Chem. Chem. Phys. 15, 11287–11298 (2013).
Hashimoto, K., Irie, H. & Fujishima, A. TiO2 photocatalysis: a historical overview and future prospects. Jpn. J. Appl. Phys. 44, 8269 (2005).
Guo, P., Schaller, R. D., Ketterson, J. B. & Chang, R. P. Ultrafast switching of tunable infrared plasmons in indium tin oxide nanorod arrays with large absolute amplitude. Nat. Photon. 10, 267–273 (2016).
Du, L., Furube, A., Hara, K., Katoh, R. & Tachiya, M. Plasmon induced electron transfer at gold–TiO2 interface under femtosecond near-IR two-photon excitation. Thin Solid Films 518, 861–864 (2009).
Wang, K., Schonbrun, E., Steinvurzel, P. & Crozier, K. B. Trapping and rotating nanoparticles using a plasmonic nano-tweezer with an integrated heat sink. Nat. Commun. 2, 469 (2011).
Chen, Y.-C., Yossifon, G. & Yang, Y.-T. Suppression of photothermal convection of microparticles in two dimensional nanoplasmonic optical lattice. Appl. Phys. Lett. 109, 201111 (2016).
Kang, Z., Chen, J., Wu, S.-Y. & Ho, H.-P. Plasmonic absorption activated trapping and assembling of colloidal crystals with non-resonant continuous gold films. RSC Adv. 5, 105409–105415 (2015).
Acknowledgements
SH acknowledges financial support from JSPS KAKENHI (Grant No. 26286004).
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Furube, A., Hashimoto, S. Insight into plasmonic hot-electron transfer and plasmon molecular drive: new dimensions in energy conversion and nanofabrication. NPG Asia Mater 9, e454 (2017). https://doi.org/10.1038/am.2017.191
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2017.191
This article is cited by
-
Design and fabrication of porous three‐dimensional Ag-doped reduced graphene oxide (3D Ag@rGO) composite for interfacial solar desalination
Scientific Reports (2024)
-
Innovative textiles treated with TiO2-AgNPs with succinic acid as a cross-linking agent for medical uses
Scientific Reports (2024)
-
Electrochemical synthesis of porous Ag@PANI/MWCNTs ternary composite for electrochemical and photocatalytic application
Polymer Bulletin (2024)
-
Nonlinear plasmonics: second-harmonic generation and multiphoton photoluminescence
PhotoniX (2023)
-
Photothermal Heating of Au Nanorods and Nanospheres: Temperature Characteristics and Strength of Convective Forces
Plasmonics (2023)
