
Abstract
Nanoparticles are among the most fascinating materials because of their unique size-dependent properties. The spatial positioning of the nanoparticles with a nanoscale precision will greatly enhance their potential for use in the fabrication of functional materials and devices. Recently, the development of DNA nanotechnology has enabled the construction of two- and three-dimensional, precisely addressable nanostructures and devices based on DNA sequence design and programmable assembly. Advances in conjugating DNA with nanoparticles can bridge these two technologies and provide scientists with a new tool to study material transportation and energy transfer at a nanometer scale. In this review, we summarize the recent progress in this emerging research field, which includes not only the static arrangements of inorganic nanoparticles but also the dynamic regulation of organic and biological units at a nanometer scale. With the rapid development in this field, new challenges and new possibilities will emerge and bring about fruitful achievements with a significant impact on future work.
Similar content being viewed by others

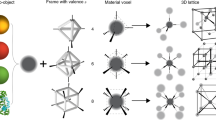
Introduction
Nanoparticles (NPs), ranging from synthetic quantum dots to natural enzymes, are fascinating materials and have attracted intense interest from both the scientific community and the public. Today, great accomplishments in the control of the size, shape, composition and dispersity of synthetic NPs, as well as the extraction, synthesis and purification of both natural and artificial biological NPs, have been achieved. The next major challenge in this field will certainly transition from studies on the individual NPs to studies on their higher-order assembly behaviors,1 which may lead to promising functions based on the interactive and collective properties of the NP ensembles. In particular, the spatial regulation of NPs with a nanoscale precision will create an opportunity to exploit and manipulate their collective properties, which is critical for many important applications in nanophotonics, nanoelectronics, nanochemistry and nanomedicine. To this end, many efforts have been made to develop top-down or bottom-up techniques to move or assemble the NPs into hierarchical structures ranging from discrete nanopatterns to periodic superlattices. However, a common problem is the extreme difficulty in achieving the nanoaddressability, dynamic tunability and continuous controllability without the need for complex instrumentation.
Meanwhile, nucleic acids have been increasingly used in materials in addition to their biological functions.2 Based on their programmable, nanoaddressable self-assembly ability and the nanosize of the DNA molecules, they have been proven to be of great utility in constructing elaborate information-encoded static nanostructures3, 4, 5 and dynamic nanomachines,6, 7 which are greatly beneficial for organizing NPs in precise manners.8 Given the pioneering work on the DNA-directed assembly of gold NPs (AuNPs) reported by Mirkin et al.9 and Alivisatos et al.10 in 1996, this field has witnessed an explosion of interest in the production of various NP patterns and ensembles with DNA templates. In addition to synthetic NPs, DNA could also direct the spatial regulation of natural NPs, such as enzymes and proteins, and therefore create a bridge between the biological and materials sciences. More importantly, although the technique is still young and evolving, the ability to easily and exquisitely regulate the assembly of NPs with DNA has already resulted in some promising applications that rely on the adjustable physical, chemical and biological properties of the ensembles.
In this review, we focus on new advances in the spatial regulation of NPs through the use of DNA nanotechnology. First, we highlight new efforts in the fabrication and application of static DNA-NP patterns, especially focusing on the research conducted after the invention of the DNA origami technique;11 this will cover two emerging topics in the regulation of plasmonics and the reactions on the DNA nanostructures. Next, the construction of DNA devices possessing designable and tunable spatial controls is illustrated, and the dynamic interactions between the organic components or the biological NPs as a function of the change in spatial distance are discussed. We also discuss the new and fascinating concept of the spatial regulation of DNA molecules on individual NPs, which may greatly enhance the construction and tailoring of NP ensembles.
Regulation of NPs by static DNA nanostructures
When all of the DNA strands in nanostructures are immobilized, this DNA nanostructure can be regarded as static. Depending on the design, static DNA nanostructures can possess varied sizes, shapes, geometries and periodicities. The most attractive feature of these nanostructures is their nanoaddressability, which creates a possibility for precise positional assembly and the immobilization of NPs into specific patterns and even crystals. With rapid developments in anchoring techniques such as DNA hybridization, bioaffinity coupling and covalent chemistry, many types of NPs (including proteins, dendrimers, metal nanocrystals and quantum dots) have been successfully assembled on DNA nanostructures. The current focus of this field has moved to exploring the potential applications of these NP patterns.12, 13 Here, we are not trying to give a comprehensive overview on the positioning methods, but instead, we are aiming to outline two emerging lines of research including the manipulations of light and reactions at the nanoscale that may lead to exciting functional nanodevices.
Formation of plasmonic patterns from metal NPs
The fast-growing field of plasmonics, which studies the abilities of metal nanostructures to localize, guide and manipulate electromagnetic waves beyond the diffraction limit and at a nanometer-length scale, is an emerging research area.14 Morphological parameters for metal nanostructures, including the size, shape, geometry and interparticle distance, could substantially influence the plasmonic coupling and the subsequent optical and electrical properties.15 Static DNA nanostructures are powerful in connecting and arranging AuNPs into spatially defined plasmonic assemblies.16
As the simplest DNA nanostructure, the single DNA duplex is easy to design and use. The interest in this field can be traced back to Mirkin’s and Alivisatos’ original work. Therefore, one focus in the study of plasmonics, based on Alivisatos’ idea, is the careful assembly of NPs with a countable number of DNA modifications into designed finite-sized shapes. Examples of this include some heterodimer model systems for the investigation of plasmonic coupling,17 chiral pyramids exhibiting R/S optical activity18 and some three-dimensional (3D) plasmonic aggregates with enhanced local fields of up to 1000 times higher than the incident field strength.19 Additionally, the assembly of fully DNA-capped metal NPs is an easy way to generate large aggregates, leading to a shift in the spectrum. In addition to the well-known color change of AuNPs shown by Mirkin et al.,9 it was recently demonstrated by Tang and co-workers20 that Au nanorod assemblies can exhibit plasmonic circular dichroism responses. More interestingly, finite-sized Fano-like metal NP assemblies can also be obtained if the length and coverage of the DNA modifications and the sizes and ratios of the NPs are well controlled.21
By increasing the complexity of DNA nanostructures, it is possible to have more exquisite control over the nanopatterning. The most attractive example may be the DNA origami technique, which can meet the critical demands of both structural robustness and nanoscale addressability from 1D to 3D.11, 22, 23, 24, 25, 26 Yan and co-workers27, 28, 29, 30 made significant contributions in this direction by establishing some reliable immobilization techniques for metal NPs on the DNA origami. Based on this advance, they reported the first DNA origami-based plasmonic nanostructure.31 This structure was created by arranging Au nanorod dimers in varying orientations and observing the plasmonic coupling-induced band shifts in the extinction spectra (Figure 1a). Other groups have focused on circular dichroism effects that have originated from the chiral geometry of AuNP arrays. Liedl and co-workers32 organized AuNPs into chirality-controlled nanohelices on a rod-like DNA origami template, resulting in defined circular dichroism and optical rotatory dispersion effects (Figure 1b). As simpler alternatives, pronounced circular dichroism is also observed from chiral AuNP (Figure 1d)33, 34 and nanorod35 arrays assembled on planar rectangular DNA origami templates. Furthermore, multiscaffold DNA origami ribbons have been shown to be powerful tools for the assembly of plasmonic waveguides consisting of linear AuNP arrays.36

Formation of plasmonic arrays from metal nanoparticles. (a) DNA origami-based plasmonic nanostructures are produced by arranging gold nanorod dimers with variable orientations. (b) The circular dichroism effect originates from the chiral geometry of the gold nanoparticle arrays. (c) Fluorescence enhancement effects are achieved with a simple DNA origami triangle as the template. (d) Circular dichroism results from the chiral gold nanoparticles arrays assembled on planar rectangular DNA origami templates.
As previously mentioned, plasmonic coupling often produces enhanced local field strengths, and such enhancements are significantly dependent on the geometry. The ability to precisely the tailor NP arrangement through the DNA origami technique may lead to the strong near-field coupling of surface plasmons and inspire new sensing techniques. For instance, if a single fluorescent dye is placed in the narrow gap of a pair of metal NPs, the fluorescence efficiency could be strongly altered by the distance between the dye and the metal NP surface. Two groups have proven that this single-molecular fluorescence enhancement effect can be observed in DNA origami-based systems by using a DNA origami pillar structure (Figure 1c)37 and a simple triangular DNA origami.38 Similarly, if Raman dyes are placed in the gap, surface-enhanced Raman scattering can be observed39 if the gap is sufficiently narrow because Raman scattering cross-sections are much smaller than fluorescence cross-sections. In a very recent work, Keyser and co-workers40 constructed a AuNP pair with a sub-5-nm gap on a multilayer DNA origami platform.40 This elegant technique allowed for the sensitive detection of dye molecules and changes in the composition of the single-stranded DNA (ssDNA) on the AuNPs. The enhancement factors were up to seven orders of magnitude as compared with bulk Raman measurements of analytes in solution.
Regulating reaction pathways for bioinspired nanofactories
In living cells, the metabolism is spatially regulated through the site-specific compartmentalization of multienzymatic cascades in subcellular organelles.41 The high level of spatial organization and integration enables the metabolism to be an optimized network of interconnected biological reactions that guide the production, transportation and consumption of nutrients and allows for the maintenance, growth and reproduction of life. Inspired by nature, synthetic biologists are aiming to build biomimetic nano/microfactories for the positional immobilization of multienzymatic cascades with nanoscale precision.42, 43 These artificial systems are expected to regulate reaction pathways at a high efficiency and thus hold great promise for producing rare chemicals, medicines and cheap renewable fuels.
The idea to use DNA templates for the spatial arrangement of enzymatic cascades was first proposed by Niemeyer et al.44 in 2001. They showed that the use of a simple DNA duplex could enhance the efficiency of many bienzymatic cascades, for example, NAD(P)H:FMN oxidoreductase and luciferase,45 glucose oxidase (GOx) and horseradish peroxidase (HRP),46 and two subdomains of cytochrome P450 BM3 in a cascade.47
Because DNA duplexes can only regulate the distance between adjacent enzymes, high-order 2D and 3D DNA nanostructures can exert more precise control on multienzymatic cascades because of their abilities to regulate not only the distance but also other parameters, such as the arrangement and the concentration. As a popular modeling system, the GOx-HRP bienzymatic cascade has been well studied with many DNA nanostructures that varied from DNA tiles,48 branched DNA structures49 and DNA origami.50, 51 All of these produced an enhanced activity. Notably, two recent works on DNA origami showed that this structure has advantages over other DNA templates and represents a promising tool for the spatial and temporal regulation of cascade pathways. In the first work, Yan and co-worker50 reported an enhancement of the GOx-HRP cascade on a rectangular DNA origami platform (Figure 2A). Of note, a protein bridge could be placed between GOx and HRP to promote an intermediate diffusion and increase the cascade efficiency. Similarly, Fu et al.51 also reported increases in the efficiency of this cascade by controlling the interenzyme distance on a rectangular DNA origami (Figure 2B). It is important to note that they developed a rapid self-assembly approach for DNA origami and constructed new 3D DNA origami nanotubes. In the nanotubes, the cascade possessed an even higher activity, implying that the confined space of a DNA nanotube may be used as a nanoscale reactor.

DNA origami-templated enzymatic cascades. (A) An enhanced glucose oxidase-horseradish peroxidase (GOx-HRP) cascade on a rectangular DNA origami. (B) Addressable nanoscale bioreactors with rapid self-assembly represent an approach for three-dimensional (3D) DNA origami nanotubes.
Additionally, while the regulation of chemical reactions between active NPs has been less frequently reported, the efforts of Gothelf and co-workers may offer some clues. At the single-molecule level, they were successful in carrying out cleavage and ligation reactions for regulating NP arrays.52 They also explored the possibility of the directed polymerization of dendrimer NPs into nanopatterns on a DNA origami template.53
Dynamic regulation of NPs with DNA
The spatial arrangements of NPs have an important role in their physical properties. Understanding how to control the dynamic regulation of their spatial arrangement is critical because it not only improves the understanding of the physical properties but also facilitates the creation of multifunctional systems. With the development of DNA motors and devices, DNA has been used to create a dynamic assembly of NPs through configurational changes of DNA in response to external stimuli. Such subtle nanoscale changes can produce precise motion control at the molecular level, and the responsiveness can be modulated with different triggers, ranging from physical cues, such as temperature, light and electricity, to chemical cues, such as ions, small molecules, DNA and proteins. The dynamic DNA nanostructures provide a new platform on which to study the multiple interactions of NPs at a nanometer scale and, more importantly, in a reversible and dynamic manner. Therefore, although this field is still in its early stages, we predict that this new area will draw increasing attention and generate tremendous excitement in the very near future.
The dynamic spatial control of NPs
In Mirkin’s pioneering work in 1996, the spatial control of NPs was discussed. Based on the thermoresponsiveness of DNA, the controlled and reversible assembly of NPs into disordered macroscopic assembly structures was achieved along with tunable optical properties. This work not only offered a new class of DNA-NP material but also developed a promising strategy to control NP assembly. During the past few decades, this method to control the aggregation of NPs has also been improved and extended to other responsive DNA systems. For instance, single-stranded DNA sequences containing a half-stretch of i-motif DNA are known to be modified onto AuNPs.54 At a high pH, DNA adopts a random coil structure, whereas at a low pH, the formation of interparticle i-motifs leads to the assembly of AuNPs into aggregates. These two states can be reversibly switched by varying the pH. In contrast, when the full stretch of i-motif DNA55 is modified onto AuNPs at a low pH, the i-motif DNA folds into a compact structure without interparticle interactions, and the AuNPs aggregate when the pH is increased.56 These examples show that by modifying the AuNPs with DNA, it is possible to reversibly switch their assembly and disassembly.
In addition to the disordered aggregates, DNA devices can also be used to control dynamically the crystallization of NPs. For example, different crystalline states of NPs have been achieved under the guidance of DNA.57, 58, 59 However, most reported superlattice structures are static. Once constructed, it is hard to control factors, such as the interparticle distance, in situ. In 2010, Maye et al.60 reported a strategy to modulate the distances in the superlattices with a reconfigurable DNA device that served as an interparticle linkage. The configuration transition among a flexible single strand, a bulged stem-loop structure and a full duplex structure of the DNA linker was achieved using the chain replacement reaction, which resulted in the in situ switching of interparticle distances in the range of 15–25%. These approaches offer opportunities for the regulation and dynamical control of NP assembly systems and hysteresis during the transition to a flexible configuration, reflecting the low level of reversibility. In 2013, Mirkin and co-workers61 proposed a new duplex design with a hairpin structure, which can be driven between the open and closed states through a similar chain replacement reaction. The introduction of stem-loop DNA molecules as small-molecule allosteric effectors maintained the crystal symmetry, but its configuration transition caused the lattice parameters to cleanly change over the 3–12 nm range. This design produced good reversibility through multiple cycles with a time scale of seconds, and this method can also be applied to different crystalline states. The strategies to control dynamically the lattices of the NPs provide the potential ability to modulate the plasmonic, catalytic and mechanical properties of this emerging class of materials.
In addition to the macroscopic assembly of the NPs, DNA motors/machines can also be used to control the spatial arrangement of NPs on a nanometer scale. For example, the distance between the Au electrode and the quantum dots was tuned in situ using the pH-sensitive i-motif DNA, resulting in a dynamic pH-driven modulation system for photoelectric conversion.62 As illustrated in Figure 3a, the Au electrode was modified with an i-motif DNA machine, and the other end of the DNA was connected with CdSe/ZnS core–shell quantum dots. With the switching between acidic and basic conditions, the i-motif machine can change its configuration and bring the quantum dots closer to/further from the Au electrode, respectively, providing a strategy to control dynamically the photoelectric conversion. Other means are also available, such as using the DNA strand displacement reaction to control the assembly of NPs and their relative positions.63, 64 As this process is dynamic, it allows experimenters to control the movement of the NPs on a designed DNA origami pattern in the desired direction.65

The spatial control of nanoparticles by DNA motors. (a) A photoelectric conversion switch based on quantum dots with i-motif DNA scaffolds. (b) DNA-molecular-motor-controlled dendron association with an i-motif structure.
In addition to controlling the spatial arrangement of inorganic NPs with DNA machines, this strategy can be applied to organic NPs and other materials. For example, two amphiphilic dendrons can be covalently connected with an i-motif DNA machine.66 As illustrated in Figure 3b, in basic conditions, the rigid double helix formed by the hybridization of DNA extends two dendrons away from each other by ~5.8 nm. As the pH decreases, the i-motif DNA machine folds into a compact quadruplex structure, bringing the two dendrons into proximity and allowing them to merge together. This process is reversible, and the association of the two dendron parts increases the stability of the i-motif DNA, whose increased melting temperature (Tm) can be used to probe the strength of the interaction between the two NPs. With the versatile modification of DNA, the dendron and protein can be bonded to both sides of the DNA, and the conformation of the dendron–DNA–protein hybrid molecular system can be controlled reversibly by the i-motif DNA machine.67 The incorporation of the DNA machine into synthetic molecules or biological components provides a new platform to achieve well-defined supramolecular assemblies, to manipulate NPs at the single molecular level in a controllable manner and to study the interactions between NPs.
Regulation of protein function through DNA motors
Proteins can be considered to be natural NPs that have important roles in both life science and material science. Since its development, DNA nanotechnology has been a good candidate for regulating the function of proteins through spatial control, and a DNA machine could enable the study of complex interactions of proteins in three dimensions. In the early stages of this field, scientists used ssDNA and double-stranded DNA to control the spatial distance.68, 69 The protein entities were covalently attached to the DNA strands. As ssDNA is usually considered flexible, while double-stranded DNA is rigid with a persistence length of ~50 nm, changes in the rigidity between ssDNA and double-stranded DNA can adjust the amount of space between two proteins. More rigid DNA structures, such as the double-crossover, Holliday junction and origami scaffolds, may provide more controllability and precision. For example, Figure 4a shows two double-crossover motifs joined with a Holliday junction forming a tweezer-like DNA machine that can be opened (in the stem-loop orientation) and closed (in the double helix orientation) by the addition of the fuel and antifuel strand, respectively.70 Two ligands can bind the target protein, thrombin, and are introduced at both terminals of the DNA tweezers. In the closed state, the two ligands are in a cooperative position and catch the thrombin. However, as the DNA tweezers open, the two ligands are forced apart, and the thrombin is released. Such a target-responsive DNA machine can be repeatedly used and provides a promising strategy to study spatially dependent interactions of NPs. In addition to controlling the arrangement of NPs, it has been reported that ‘DNA origami pliers’ and ‘DNA origami forceps’ containing ~170-nm-long levers connected by a fulcrum can be used to detect targets ranging from metal ions to proteins in a single molecular lever.71 This is accomplished by observing the shape transition of the DNA origami with atomic force microscopy.

Regulation of protein binding affinity. (a) The reversible regulation of a protein binding affinity by a DNA machine (b) A DNA tweezer-actuated enzyme nanoreactor.
In living systems, a series of enzymatic reactions occurs via cascades. The efficiencies and specificities of the enzymatic cascade reactions rely on the appropriate spatial arrangement. DNA nanotechnology allows for the predictable and programmable positioning of the NPs in a precise manner. By taking advantage of this fact, previous studies have shown that it is feasible to locate multiple enzymes on static DNA nanostructures. However, the distance between enzymes was determined and fixed by the underlying DNA structures. Therefore, to mimic the enzyme cascade reaction in vitro, where the enzyme locations are not consistent, scientists have turned to dynamic DNA nanostructures. Following the use of DNA tweezers to regulate protein binding, a similar design has been used to attach two enzymes of GOx and HRP to both arm ends of DNA tweezers.72 As this reaction occurs, the diffusion distance of intermediate H2O2 determines the rate of this enzymatic cascade reaction. By deliberately tuning the state of DNA tweezers in closed and open states, the distance between GOx and HRP can be adjusted from several nanometers to ~18 nm. In the closed state, GOx and HRP are brought into proximity, leading to a much closer diffusion pathway for H2O2. In contrast, in the open state, both enzymes are spatially separated, thus lowering their enzymatic efficiency. It has been shown that several cycles of regulation of this enzyme cascade reaction can be successfully conducted. Furthermore, a proof of concept of this idea was also confirmed in another system to actuate the activity of an enzyme/cofactor pair.73 A dehydrogenase and NAD+ cofactor are fixed on the DNA tweezers, and the enzymatic inhibition and activation can be reversibly turned on and off upon the tuning of the open and closed state of the DNA tweezers, respectively (Figure 4b). This approach allows for the dynamic regulation of other enzymatic systems and the replication of enzyme cascades outside of living organisms.
Spatial regulation of DNA modifications on NPs
There has been a recent increase in interest in the study of NPs with discrete and/or asymmetric surface modifications, such as patchy NPs. The introduced surface modifications break up the original symmetry of the NPs and facilitate their subsequent higher-order self-assembly in a designed manner. Famous examples can be found in the landmark work of Alivisatos on controlling the number of DNA modifications on metal NPs and quantum dots and on using these DNA-NPs for building discrete ensembles. Now, new challenges have emerged in regulating both the position and density of the DNA modifications on not only the symmetric but also the asymmetric NPs. This is also a critical issue for the regulation of DNA modifications on organic or natural NPs.
DNA-inorganic NPs
AuNPs are one of the most important inorganic NPs.74 One critical remaining challenge is to control the position and density of the DNA modifications on the AuNP surface.75 Positional control aims to achieve the addressable surface modification on the AuNPs, either through symmetric/isotropic or asymmetric/anisotropic modifications. The density control, in contrast, aims to modify the AuNPs with an exact number of DNA strands and to control the surface coverage density on a AuNP with uniform DNA modifications. In principle, such spatial control could not only increase the precision of the positioning assembly but also improve the hybridization efficiency.
Early efforts were based on the careful control of the stoichiometry with subsequent postsynthesis separation.75 Discrete DNA-AuNPs with an exact number of DNA modifications could be obtained, although the yield was low. To overcome this issue, new ideas have been developed to use the steric hindrance. In this approach, an assisted particle or molecule with a size larger than the target AuNP is preattached to the surface to block the potential DNA-anchoring sites. Thus, only a small area, normally on the surface opposite to the hindrance-blocked surface, on the AuNP is free to be modified with DNA. Up to sixfold symmetric DNA modifications on AuNPs have been reported using this strategy.76 If a structured DNA motif rather than a simple ssDNA strand is used,77 a dual steric hindrance effect is produced. Reports have shown that the yield of mono-DNA modifications could be increased to 70% in such a setup (Figure 5a).78 One advantage of this high-yield modification is that it can simplify the postsynthesis separation. The positional control of DNA modifications could also be partially solved with the help of a geometric template with many anchors. For instance, a DNA duplex with two adjustable thiol groups was reported for the hetero-bimodification on an AuNP.79 In another case, a simple DNA duplex with two thiol labels at the same end was used for the bimodification of the same site on an AuNP.80 Of note, one end of the DNA was attached to the AuNP through its 5′ end and the other through its 3′ end, making the conjugates unidirectional.

DNA–AuNP conjugates. (a) Preparation of mono-DNA modifications on AuNPs by combining both DNA and microspheres in a dual steric hindrance. (b) Spatially isolated and highly hybridizable functionalization of gold nanoparticles with diblock oligonucleotides.
However, precisely adjusting the surface DNA density while simultaneously allowing the attached DNA strands to adopt upright conformations is very difficult. In a recent work reported by Pei and co-workers,81 a new non-thiolated DNA modification strategy was proposed in which a unique polyA block could be specifically absorbed on the AuNP surface while acting as a hindrance to force the rest of the sequence to be upright (Figure 5b). The upright conformation can avoid the nonspecific absorption of DNA on AuNPs and can speed up the hybridization behavior of DNA-AuNPs.
Apart from AuNPs, the conjugation of DNA with other noble metal NPs and quantum dots can be obtained. Because these NPs are not as stable as AuNPs, the capping strategy must be optimized. The thiol approach is still suitable for PtNPs,82 but phosphorothioate linkages also have been used for AgNPs83 as well as for some quantum dots.84
DNA–protein NPs
The conjugation, especially the site-specific conjugation, of DNA to proteins or peptides is another important issue85 and may be promising for bio-orthogonality.86 There are two major strategies: chemical conjugation and biological coupling. Chemical conjugation is traditionally and widely used for bioconjugation.87 Typical bioconjugate chemistry includes amine reactions, thiol reactions and ‘click’ reactions. Proteins or peptides can be covalently coupled to DNA strands directly or using homo- or hetero-multifunctional linkers. It is important to achieve site-specific coupling on both the DNA strand and the protein surface. Hocek and co-workers recently reported the incorporation of vinylsulfonamide or acrylamide-modified cytosine in the process of prime extension. Therefore, the cysteine-containing peptide could be coupled to the DNA strand site specifically on cytosine sites.88 Another advantage of this technique is that these groups are more stable than the commonly used maleimide. Using a similar strategy, these authors also reported the conjugation between aldehyde–DNA and lysine–peptide.89
The chemical conjugation strategy uses strong covalent bonds to conjugate DNA and protein molecules. However, this strategy has difficulty in meeting the increasing requirements of reversibility, simplicity and biocompatibility. Therefore, biological coupling methods, such as bioaffinity coupling or enzymatic ligation, are important alternatives. The biotin–streptavidin, His-tag and apoenzyme–cofactor interaction pairs are all commonly used for bioaffinity coupling. Comparatively, enzymatic ligation90 is an emerging strategy for DNA–protein conjugation. For instance, a specific enzyme Gene-A* protein can site selectively cleave its recognition sequence and then couple to the 5′ end of the DNA sequence.91 Terminal deoxynucleotidyl transferase has been used for the direct enzymatic ligation of native DNA (through the 3′ end) to nucleotide triphosphates coupled to proteins and other large macromolecules.92 In another interesting work,93 Tao and Zhu93 built an in vitro translation system in which the newly synthesized protein or peptide could be coupled in situ to a puromycin-modified ssDNA. This method has been found in applications for the in situ protein chip fabrication and in vitro display.94
Perspectives
Although various methods of combining DNA and NPs have been established, the development of new conjugation strategies to achieve spatially controlled and anisotropic conjugation on NPs remains a challenge. Additionally, further improvements in the methods of conjugating DNA with biological components are required. Such biological molecules often contain multiple surface functional groups that must connect to the DNA at specific sites without changing its biological functions. In terms of the static nanostructure of DNA-NPs, several aspects will require further investigation. First, artificial bionanoreactors, which are DNA–biomolecule conjugates anchored on spatially addressable DNA templates to produce controlled cascades, could be used to mimic in vivo bioreactions in compartments. Second, addressable nanocircuits, which are DNA–metal NPs assembled on DNA origami templates to form nanocircuits, could be used to overcome the limits of optical lithography. Third, nanophotonic devices, which are also DNA–metal NPs assembled on 2D/3D DNA templates to form metal NP arrays with controlled plasmonic modes, are also promising.
Another area of interest is developing structures from the nano- to the microscale or even macroscale using DNA–NP conjugates as artificial atoms for the construction of finite or infinite, periodic or aperiodic large structures. With the success of constructing DNA motors or machines, spatially controlling the distance of NPs is becoming feasible. This reversible regulation enables the in situ study of the distance-dependent interactions between the NPs. Although this model system is simple and only in the proof-of-concept stage, future work will focus on the construction of dynamic structures for the incorporation of multiple NPs and will allow the study of complex and multiple interactions. The most challenging aspect will be to continuously and precisely control the spatial distance at the nanoscale, which will provide a new platform for mimicking polyvalent interactions in vitro. In the long term, the cost of DNA is an issue. So far, the DNA for constructing nanostructures and conjugating with NPs is programmed and synthesized, lowering the cost and synthesis at a large scale. This will promote the application of DNA-NPs. Moreover, the quality of DNA structures combined with NPs and their uniformity, stability and toxicity must be improved in future studies. With the rapid development of DNA nanotechnology, the regulation of DNA-NPs by static and dynamic nanostructures will achieve tremendous success.
References
Nie, Z., Petukhova, A. & Kumacheva, E Properties and emerging applications of self-assembled structures made from inorganic nanoparticles. Nat. Nanotechnol. 5, 15–25 (2010).
Seeman, N. C DNA in a material world. Nature 421, 427–431 (2003).
Seeman, N. C Nanomaterials based on DNA. Annu. Rev. Biochem. 79, 65–87 (2010).
Ke, Y., Ong, L. L., Shih, W. M. & Yin, P Three-dimensional structures self-assembled from DNA bricks. Science 338, 1177–1183 (2012).
Wei, B., Dai, M. & Yin, P Complex shapes self-assembled from single-stranded DNA tiles. Nature 485, 623–626 (2012).
Liu, H. & Liu, D DNA nanomachines and their functional evolution. Chem. Commun. 2625–2636 (2009).
Liu, D., Cheng, E. & Yang, Z DNA-based switchable devices and materials. NPG Asia Mater. 3, 109–114 (2011).
Aldaye, F. A., Palmer, A. L. & Sleiman, H. F Assembling materials with DNA as the guide. Science 321, 1795–1799 (2008).
Mirkin, C. A., Letsinger, R. L., Mucic, R. C. & Storhoff, J. J A DNA-based method for rationally assembling nanoparticles into macroscopic materials. Nature 382, 607–609 (1996).
Alivisatos, A. P., Johnsson, K. P., Peng, X., Wilson, T. E., Loweth, C. J., Bruchez, M. P. & Schultz, P. G Organization of 'nanocrystal molecules' using DNA. Nature 382, 609–611 (1996).
Rothemund, P. W. K Folding DNA to create nanoscale shapes and patterns. Nature 440, 297–302 (2006).
Pinheiro, A. V., Han, D., Shih, W. M. & Yan, H Challenges and opportunities for structural DNA nanotechnology. Nat. Nanotechnol. 6, 763–772 (2011).
Wang, Z. -G. & Ding, B DNA-based self-assembly for functional nanomaterials. Adv. Mater. 25, 3905–3914 (2013).
Gramotnev, D. K. & Bozhevolnyi, S. I Plasmonics beyond the diffraction limit. Nat. Photon 4, 83–91 (2010).
Rycenga, M., Cobley, C. M., Zeng, J., Li, W., Moran, C. H., Zhang, Q., Qin, D. & Xia, Y Controlling the synthesis and assembly of silver nanostructures for plasmonic applications. Chem. Rev. 111, 3669–3712 (2011).
Tan, S. J., Campolongo, M. J., Luo, D. & Cheng, W Building plasmonic nanostructures with DNA. Nat. Nanotechnol. 6, 268–276 (2011).
Sheikholeslami, S., Jun, Y.-w., Jain, P. K. & Alivisatos, A. P Coupling of optical resonances in a compositionally asymmetric plasmonic nanoparticle dimer. Nano Lett. 10, 2655–2660 (2010).
Yan, W., Xu, L., Xu, C., Ma, W., Kuang, H., Wang, L. & Kotov, N. A Self-assembly of chiral nanoparticle pyramids with strong R/S optical activity. J. Am. Chem. Soc. 134, 15114–15121 (2012).
Barrow, S. J., Wei, X., Baldauf, J. S., Funston, A. M. & Mulvaney, P The surface plasmon modes of self-assembled gold nanocrystals. Nat. Commun. 3, 1275 (2012).
Li, Z., Zhu, Z., Liu, W., Zhou, Y., Han, B., Gao, Y. & Tang, Z Reversible plasmonic circular dichroism of Au nanorod and DNA assemblies. J. Am. Chem. Soc. 134, 3322–3325 (2012).
Fan, J. A., He, Y., Bao, K., Wu, C., Bao, J., Schade, N. B., Manoharan, V. N., Shvets, G., Nordlander, P., Liu, D. R. & Capasso, F DNA-enabled self-assembly of plasmonic nanoclusters. Nano Lett. 11, 4859–4864 (2011).
Douglas, S. M., Dietz, H., Liedl, T., Högberg, B., Graf, F. & Shih, W. M Self-assembly of DNA into nanoscale three-dimensional shapes. Nature 459, 414–418 (2009).
Dietz, H., Douglas, S. M. & Shih, W. M Folding DNA into twisted and curved nanoscale shapes. Science 325, 725–730 (2009).
Han, D., Pal, S., Nangreave, J., Deng, Z., Liu, Y. & Yan, H DNA origami with complex curvatures in three-dimensional space. Science 332, 342–346 (2011).
Sobczak, J.-P. J., Martin, T. G., Gerling, T. & Dietz, H Rapid folding of DNA into nanoscale shapes at constant temperature. Science 338, 1458–1461 (2012).
Han, D., Pal, S., Yang, Y., Jiang, S., Nangreave, J., Liu, Y. & Yan, H DNA gridiron nanostructures based on four-arm junctions. Science 339, 1412–1415 (2013).
Sharma, J., Chhabra, R., Andersen, C. S., Gothelf, K. V., Yan, H. & Liu, Y Toward reliable gold nanoparticle patterning on self-assembled DNA nanoscaffold. J. Am. Chem. Soc. 130, 7820–7821 (2008).
Ding, B., Deng, Z., Yan, H., Cabrini, S., Zuckermann, R. N. & Bokor, J Gold nanoparticle self-similar chain structure organized by DNA origami. J. Am. Chem. Soc. 132, 3248–3249 (2010).
Pal, S., Deng, Z., Ding, B., Yan, H. & Liu, Y DNA-origami-directed self-assembly of discrete silver-nanoparticle architectures. Angew. Chem. Int. Ed. 49, 2700–2704 (2010).
Zhao, Z., Jacovetty, E. L., Liu, Y. & Yan, H Encapsulation of gold nanoparticles in a DNA origami cage. Angew. Chem. Int. Ed. 50, 2041–2044 (2010).
Pal, S., Deng, Z., Wang, H., Zou, S., Liu, Y. & Yan, H DNA directed self-assembly of anisotropic plasmonic nanostructures. J. Am. Chem. Soc. 133, 17606–17609 (2011).
Kuzyk, A., Schreiber, R., Fan, Z., Pardatscher, G., Roller, E. M., Högele, A., Simmel, F. C., Govorov, A. O. & Liedl, T DNA-based self-assembly of chiral plasmonic nanostructures with tailored optical response. Nature 483, 311–314 (2012).
Shen, X., Song, C., Wang, J., Shi, D., Wang, Z., Liu, N. & Ding, B Rolling up gold nanoparticle-dressed DNA origami into three-dimensional plasmonic chiral nanostructures. J. Am. Chem. Soc. 134, 146–149 (2012).
Shen, X., Asenjo-Garcia, A., Liu, Q., Jiang, Q., García de Abajo, F. J., Liu, N. & Ding, B Three-dimensional plasmonic chiral tetramers assembled by DNA origami. Nano Lett. 13, 2128–2133 (2013).
Lan, X., Chen, Z., Dai, G., Lu, X., Ni, W. & Wang, Q Bifacial DNA origami-directed discrete, three-dimensional, anisotropic plasmonic nanoarchitectures with tailored optical chirality. J. Am. Chem. Soc. 135, 11441–11444 (2013).
Klein, W. P., Schmidt, C. N., Rapp, B., Takabayashi, S., Knowlton, W. B., Lee, J., Yurke, B., Hughes, W. L., Graugnard, E. & Kuang, W Multiscaffold DNA origami nanoparticle waveguides. Nano Lett. 13, 3850–3856 (2013).
Acuna, G. P., Möller, F. M., Holzmeister, P., Beater, S., Lalkens, B. & Tinnefeld, P Fluorescence enhancement at docking sites of DNA-directed self-assembled nanoantennas. Science 338, 506–510 (2012).
Pal, S., Dutta, P., Wang, H., Deng, Z., Zou, S., Yan, H. & Liu, Y. Quantum efficiency modification of organic fluorophores using gold nanoparticles on DNA origami scaffolds. J. Phys. Chem. C 117, 12735–12744 (2013).
Prinz, J., Schreiber, B., Olejko, L., Oertel, J., Rackwitz, J., Keller, A. & Bald, I DNA origami substrates for highly sensitive surface-enhanced Raman scattering. J. Phys. Chem. Lett. 4, 4140–4145 (2014).
Thacker, V. V., Herrmann, L. O., Sigle, D. O., Zhang, T., Liedl, T., Baumberg, J. J. & Keyser, U. F DNA origami based assembly of gold nanoparticle dimers for surface-enhanced Raman scattering. Nat. Commun. 5, 3448 (2014).
Agapakis, C. M., Boyle, P. M. & Silver, P. A Natural strategies for the spatial optimization of metabolism in synthetic biology. Nat. Chem. Biol. 8, 527–535 (2012).
Chen, A. H. & Silver, P. A Designing biological compartmentalization. Trends Cell Biol. 22, 662–670 (2012).
Schoffelen, S. & van Hest, J. C. M Chemical approaches for the construction of multi-enzyme reaction systems. Curr. Opin. Struct. Biol. 23, 613–621 (2013).
Niemeyer, C. M Bioorganic applications of semisynthetic DNA–protein conjugates. Chem. Eur. J. 7, 3189–3195 (2001).
Niemeyer, C. M., Koehler, J. & Wuerdemann, C DNA-directed assembly of bienzymic complexes from in vivo biotinylated NAD(P)H:FMN oxidoreductase and luciferase. ChemBioChem 3, 242–245 (2002).
Müller, J. & Niemeyer, C. M DNA-directed assembly of artificial multienzyme complexes. Biochem. Biophys. Res. Commun. 377, 62–67 (2008).
Erkelenz, M., Kuo, C.-H. & Niemeyer, C. M DNA-mediated assembly of cytochrome P450 BM3 subdomains. J. Am. Chem. Soc. 133, 16111–16118 (2011).
Wilner, O. I., Weizmann, Y., Gill, R., Lioubashevski, O., Freeman, R. & Willner, I Enzyme cascades activated on topologically programmed DNA scaffolds. Nat. Nanotechnol. 4, 249–254 (2009).
Liu, Y., Du, J., Yan, M., Lau, M. Y., Hu, J., Han, H., Yang, O. O., Liang, S., Wei, W., Wang, H., Li, J., Zhu, X., Shi, L., Chen, W., Ji, C. & Lu, Y Biomimetic enzyme nanocomplexes and their use as antidotes and preventive measures for alcohol intoxication. Nat. Nanotechnol. 8, 187–192 (2013).
Fu, J., Liu, M., Liu, Y., Woodbury, N. W. & Yan, H Interenzyme substrate diffusion for an enzyme cascade organized on spatially addressable DNA nanostructures. J. Am. Chem. Soc. 134, 5516–5519 (2012).
Fu, Y., Zeng, D., Chao, J., Jin, Y., Zhang, Z., Liu, H., Li, D., Ma, H., Huang, Q., Gothelf, K. V. & Fan, C Single-step rapid assembly of DNA origami nanostructures for addressable nanoscale bioreactors. J. Am. Chem. Soc. 135, 696–702 (2013).
Voigt, N. V., Tørring, T., Rotaru, A., Jacobsen, M. F., Ravnsbaek, J. B., Subramani, R., Mamdouh, W., Kjems, J., Mokhir, A., Besenbacher, F. & Gothelf, K. V Single-molecule chemical reactions on DNA origami. Nat. Nanotechnol. 5, 200–203 (2010).
Liu, H., Torring, T., Dong, M., Rosen, C. B., Besenbacher, F. & Gothelf, K. V DNA-templated covalent coupling of G4 PAMAM dendrimers. J. Am. Chem. Soc. 132, 18054–18056 (2010).
Wang, W., Liu, H., Liu, D., Xu, Y., Yang, Y. & Zhou, D Use of the interparticle i-motif for the controlled assembly of gold nanoparticles. Langmuir 23, 11956–11959 (2007).
Liu, D. & Balasubramanian, S A proton-fuelled DNA nanomachine. Angew. Chem. Int. Ed. 42, 5734–5736 (2003).
Sharma, J., Chhabra, R., Yan, H. & Liu, Y pH-driven conformational switch of ‘i-motif’ DNA for the reversible assembly of gold nanoparticles. Chem. Commun. 477–479 (2007).
Nykypanchuk, D., Maye, M. M., van der Lelie, D. & Oleg, G DNA-guided crystallization of colloidal nanoparticles. Nature 451, 549–552 (2008).
Park, S. Y., Lytton-Jean, A. K. R., Lee, B., Weigand, S., Schatz, G. C. & Mirkin, C. A DNA-programmable nanoparticle crystallization. Nature 451, 553–556 (2008).
Macfarlane, R. J., Lee, B., Jones, M. R., Harris, N., Schatz, G. C. & Mirkin, C. A Nanoparticle superlattice engineering with DNA. Science 334, 204–208 (2011).
Maye, M. M., Kumara, M. T., Nykypanchuk, D., Sherman, W. B. & Gang, O Switching binary states of nanoparticle superlattices and dimer clusters by DNA strands. Nat. Nanotechnol. 5, 116–120 (2010).
Kim, Y., Macfarlane, R. J. & Mirkin, C. A Dynamically interchangeable nanoparticle superlattices through the use of nucleic acid-based allosteric effectors. J. Am. Chem. Soc. 135, 10342–10345 (2013).
Meng, H., Yang, Y., Chen, Y., Zhou, Y., Liu, Y., Chen, X., Ma, H., Tang, Z., Liu, D. & Jiang, L Photoelectric conversion switch based on quantum dots with i-motif DNA scaffolds. Chem. Commun. 2293–2295 (2009).
Song, T. & Liang, H Synchronized assembly of gold nanoparticles driven by a dynamic DNA-fueled molecular machine. J. Am. Chem. Soc. 134, 10803–10806 (2012).
Elbaz, J., Cecconello, A., Fan, Z., Govorov, A. O. & Willner, I Powering the programmed nanostructure and function of gold nanoparticles with catenated DNA machines. Nat. Commun. 4, 2000 (2013).
Gu, H., Chao, J., Xiao, S.-J. & Seeman, N. C A proximity-based programmable DNA nanoscale assembly line. Nature 465, 202–205 (2010).
Sun, Y., Liu, H., Xu, L., Wang, L., Fan, Q. & Liu, D DNA-molecular-motor-controlled dendron association. Langmuir 26, 12496–12499 (2010).
Sun, Y., Liu, H., Xu, L., Wang, L., Fan, Q. & Liu, D A pH responsive dendron–DNA–protein hybrid supramolecular system. Soft Matter 6, 2143–2145 (2010).
Röglin, L., Ahmadian, M. R. & Seitz, O DNA-controlled reversible switching of peptide conformation and bioactivity. Angew. Chem. Int. Ed. 46, 2704–2707 (2007).
Williams, A. R., Diehnelt, C. W., Belcher, P., Greving, M., Woodbury, N. W. & Johnston, S. A Creating protein affinity reagents by combining peptide ligands on synthetic DNA scaffolds. J. Am. Chem. Soc. 131, 17233–17241 (2009).
Zhou, C., Yang, Z. & Liu, D Reversible regulation of protein binding affinity by a DNA machine. J. Am. Chem. Soc. 134, 1416–1418 (2012).
Kuzuya, A., Sakai, Y., Yamazaki, T., Xu, Y. & Komiyama, M Nanomechanical DNA origami 'single-molecule beacons' directly imaged by atomic force microscopy. Nat. Commun. 2, 449 (2011).
Xin, L., Zhou, C., Yang, Z. & Liu, D Regulation of an enzyme cascade reaction by a DNA machine. Small 9, 3088–3091 (2013).
Liu, M., Fu, J., Hejesen, C., Yang, Y., Woodbury, N. W., Gothelf, K., Liu, Y. & Yan, H A DNA tweezer-actuated enzyme nanoreactor. Nat. Commun. 4, 2127 (2013).
Saha, K., Agasti, S. S., Kim, C., Li, X. & Rotello, V. M Gold nanoparticles in chemical and biological sensing. Chem. Rev. 112, 2739–2779 (2012).
Zhang, T., Yang, Z. & Liu, D DNA discrete modified gold nanoparticles. Nanoscale 3, 4015–4021 (2011).
Kim, J.-W., Kim, J.-H. & Deaton, R DNA-linked nanoparticle building blocks for programmable matter. Angew. Chem. Int. Ed. 50, 9185–9190 (2011).
Zhang, T., Chen, P., Sun, Y., Xing, Y., Yang, Y., Dong, Y., Xu, L., Yang, Z. & Liu, D A new strategy improves assembly efficiency of DNA mono-modified gold nanoparticles. Chem. Commun. 47, 5774–5776 (2011).
Li, Z., Cheng, E., Huang, W., Zhang, T., Yang, Z., Liu, D. & Tang, Z Improving the yield of mono-DNA-functionalized gold nanoparticles through dual steric hindrance. J. Am. Chem. Soc. 133, 15284–15287 (2011).
Suzuki, K., Hosokawa, K. & Maeda, M Controlling the number and positions of oligonucleotides on gold nanoparticle surfaces. J. Am. Chem. Soc. 131, 7518–7519 (2009).
Li, Z., Cheng, E., Huang, W., Zhang, T., Yang, Z., Liu, D. & Tang, Z DNA bimodified gold nanoparticles. Langmuir 28, 1966–1970 (2011).
Li, Z., Cheng, E., Huang, W., Zhang, T., Yang, Z., Liu, D. & Tang, Z Designed diblock oligonucleotide for the synthesis of spatially isolated and highly hybridizable functionalization of DNA-gold nanoparticle nanoconjugates. J. Am. Chem. Soc. 134, 11876–11879 (2012).
Wang, L., Feng, Y., Yang, Z., He, Y., Fan, Q. & Liu, D Reversibly controlled morphology transformation of an amphiphilic DNA–dendron hybrid. Chem. Commun. 48, 3715–3717 (2012).
Zheng, Y., Li, Y. & Deng, Z Silver nanoparticle–DNA bionanoconjugates bearing a discrete number of DNA ligands. Chem. Commun. 48, 6160–6162 (2012).
Tikhomirov, G., Hoogland, S., Lee, P. E., Fischer, A., Sargent, E. H. & Kelley, S. O DNA-based programming of quantum dot valency, self-assembly and luminescence. Nat. Nanotechnol. 6, 485–490 (2011).
Niemeyer, C. M Semisynthetic DNA–protein conjugates for biosensing and nanofabrication. Angew. Chem. Int. Ed. 49, 1200–1216 (2010).
Sletten, E. M. & Bertozzi, C. R Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 48, 6974–6998 (2009).
Hermanson, G. T. Bioconjugate Techniques, 2nd edn (Academic Press, New York, NY, USA, 2008).
Jitka, D., Petr, O., Radek, P., Marie, B., Miroslav, F. & Michal, H Vinylsulfonamide and acrylamide modification of DNA for crosslinking with proteins. Angew. Chem. Int. Ed. 52, 10515–10518 (2013).
Raindlova, V., Pohl, R. & Hocek, M Synthesis of aldehyde-linked nucleotides and DNA and their bioconjugations with lysine and peptides through reductive amination. Chem. Eur. J. 18, 4080–4087 (2012).
Sunbul, M. & Yin, J Site specific protein labeling by enzymatic posttranslational modification. Org. Biomol. Chem. 7, 3361–3371 (2009).
Mashimo, Y., Maeda, H., Mie, M. & Kobatake, E Construction of semisynthetic DNA–protein conjugates with phi X174 gene-A* protein. Bioconjugate Chem. 23, 1349–1355 (2012).
Jitka, D., Petr, O., Radek, P., Marie, B., Miroslav, F. & Michal, H Enzymatic ligation of large biomolecules to DNA. ACS Nano 7, 8098–8104 (2013).
Tao, S.-C. & Zhu, H Protein chip fabrication by capture of nascent polypeptides. Nat. Biotechnol. 24, 1253–1254 (2006).
Ishizawa, T., Kawakami, T., Reid, P. C. & Murakami, H TRAP display: a high-speed selection method for the generation of functional polypeptides. J. Am. Chem. Soc. 135, 5433–5440 (2013).
Acknowledgements
We thank the National Basic Research Program of China (973 program, No. 2013CB932803), the National Natural Science Foundation of China (Nos. 91427302, 21421064, 21174077, 21474059), the NSFC-DFG joint project TRR61 and the Beijing Municipal Science and Technology Commission for their financial support.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Yang, Z., Liu, H. & Liu, D. Spatial regulation of synthetic and biological nanoparticles by DNA nanotechnology. NPG Asia Mater 7, e161 (2015). https://doi.org/10.1038/am.2015.2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2015.2
This article is cited by
-
Soft 2D nanoarchitectonics
NPG Asia Materials (2018)
