
Abstract
Although many oxide semiconductors possess wide bandgaps in the ultraviolet (UV) regime, currently the majority of them cannot efficiently emit UV light because the band-edge optical transition is forbidden in a perfect lattice as a result of the symmetry of the band-edge states. This quantum mechanical rule severely constrains the optical applications of wide-bandgap oxides, which is also the reason why so few oxides enjoy the success of ZnO. Here, using SnO2 as an example, we demonstrate both theoretically and experimentally that UV photoluminescence and electroluminescence can be recovered and enhanced in wide-bandgap oxide thin films with ‘forbidden’ energy gaps by engineering their nanocrystalline structures. In our experiments, the tailored low-temperature annealing process results in a hybrid structure containing SnO2 nanocrystals in an amorphous matrix, and UV emission is observed in such hybrid SnO2 thin films, indicating that the quantum mechanical dipole-forbidden rule has been effectively overcome. Using this approach, we demonstrate the first prototypical electrically pumped UV-light-emitting diode based on nanostructured SnO2 thin films.
Similar content being viewed by others

Introduction
Exploitation of wide-bandgap oxides has been a time-enduring research theme as a result of their rich functionalities and technological applications.1, 2, 3, 4, 5 Among the functional oxides, tin dioxide (SnO2), is extensively applied in the fields of gas sensors, transparent conducting thin films, catalysis, solar cells and so on due to its excellent optical and electrical properties.6, 7, 8, 9, 10, 11, 12, 13 Although SnO2 has a wide direct bandgap of ∼3.6 eV (ref. 12) as well as a large exciton-binding energy of ∼130 meV,14 which compare favorably to ZnO (60 meV) and GaN (25 meV), it is commonly believed that SnO2 is not a suitable ultraviolet (UV) light emitter due to the dipole-forbidden nature of its band-edge quantum states.15 The even-parity symmetry of the conduction-band minimum and valence-band maximum states in SnO2 prohibits the band-edge radiative transition and light emission,16 which has hindered its potential use in optical applications, such as photodetectors and light-emitting diodes (LEDs). There has been much less work on the optical properties of SnO2 than on other wide-bandgap oxides such as ZnO. Most of the reports in literature suggested a dominant broad photoluminescence (PL) peak in the visible range,17, 18, 19, 20, 21, 22, 23, 24, 25 whereas others revealed the existence of UV emission in SnO2.26, 27, 28, 29, 30, 31 Thin films are indispensable for device applications, but there have been only a few works so far on the optical properties of SnO2 thin films.32, 33 Until now, light emission in SnO2 and the related mechanisms have remained as an issue of debates, and there is also a paramount need for theoretical insights. Furthermore, neither electroluminescence (EL) in the UV regime nor LEDs have ever been demonstrated in SnO2 thin films.
To reliably recover the optical activity of oxides with ‘forbidden’ bandgaps, including SnO2, and use them as UV light emitters, we need to modify their band structures, in particular the symmetry of band-edge quantum states. It is expected that the band structure and the associated optical transitions in nanostructured materials can be significantly different from those of the bulk counterparts, as they can no longer be described as infinite periodic lattice systems, invalidating the assumption of the Bloch theorem. Furthermore, such nanostructured materials are characterized by high surface-to-volume ratios, which often bring about defect-induced electronic states near the band edges. Although conventional bulks and thin films also have surfaces, nanostructured materials apparently are more efficient to break the ideal translation symmetry. However, there has been no complementary experimental and theoretical investigation so far to elucidate the correlation between the emerging light-emitting properties and the altered symmetry of band-edge states in such nanomaterials.
Here we address the challenge of designing and synthesizing UV-light-emitting SnO2-based thin films via nanostructure engineering and local symmetry breaking. Our results suggest that nanostructuring can bring about the benefits of breaking the bulk dipole-forbidden rule and effectively recovering the light emission in materials with ‘forbidden’ energy gaps. We show that a facile annealing process can controllably tune the crystallization of amorphous SnO2 films, and the resulting nanocrystal surfaces and defects have important roles in realizing the UV light emission. Furthermore, we fabricate and demonstrate the first electrically pumped LED based on nanocrytal-containing SnO2 thin films.
Materials and methods
First-principles calculations
The electronic properties of SnO2 were calculated using the density functional theory as implemented in the Vienna ab-initio simulation package code. Standard frozen-core projector augmented-wave method34, 35 and the generalized gradient approximation (GGA) to the exchange-correlation functional were used. The kinetic energy cutoff for basis functions is 400 eV. The optical properties were calculated from the complex dielectric function using the projector augmented-wave method,36 which is based on the single-electron approximation. A detailed setup of the calculations can be found in the Supplementary information.
Thin film fabrication and characterization
SnO2 thin films with a thickness of 300 nm were fabricated on quartz, p-GaN/sapphire and MgO (∼15 nm)/p-GaN/sapphire substrates at room temperature using pulsed laser deposition. The SnO2/quartz samples were annealed at 400, 600 or 800 °C in air for 30 min to controllably crystallize the amorphous SnO2 thin films. For structural characterizations, X-ray diffraction was carried out using a Bruker D8 Advance powder diffractometer (Bruker, Karlsruhe, Germany) with Cu Kα radiation. Raman and PL measurements were performed using a He–Cd laser with a 325-nm line as the excitation source. A high-resolution transmission electron microscope (JEOL 2100, Tokyo, Japan) was used to examine the crystalline structure and the nanocrystal size distribution in the hybrid thin films.
Device fabrication
For the EL experiments, the SnO2/p-GaN and the SnO2/MgO/p-GaN heterojunction were first annealed at 400 °C, and then Ni/Au electrodes were deposited through a shadow mask on the p-GaN layer. On the SnO2 layer, an ITO layer with a thickness of ∼100 nm was first grown by sputtering, and then melt In metal served as the n-type electrode. As the band structure of Mg-doped GaN wafers depends on the Mg concentration and the excitation intensity, we made sure that our samples are fabricated using substrates cut from the same wafer whose PL emission peaks at ∼445 nm (2.8 eV), and the PL excitation intensity was fixed at 0.2 W cm−2. The resulting structure of the EL devices can be found in the Supplementary information.
Results and discussion
We first used first-principles calculations to elucidate the general quantum mechanical relationship between the dipole-forbidden transition rule and the optical interband transition spectrum. Regarding the calculation method, it is well known that GGA severely underestimates the bandgap of oxide materials. However, because GGA does not significantly change the wavefunction characters at the band edges, the characters of the near-band-edge optical transitions are not affected by the GGA bandgap error.
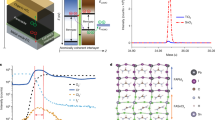
Figure 1 illustrates the concept that guided our experimental design of light-emitting nanostructured SnO2 thin films. In any crystal structure with an inversion center, optical transition is only allowed between states with opposite parity, as the electric-dipole operator is of odd parity. For bulk SnO2 with a rutile crystalline structure, both the conduction-band minimum and the valence-band maximum states occur at the Γ point with even parity; therefore, as shown in Figure 1a, the onset of the polarized optical absorption edge (denoted as αzz and αxy) is much higher than the fundamental bandgap. In other words, the band-edge transition is not allowed in ideal SnO2. To illustrate the concept of band engineering in nanomaterials, we truncated the SnO2 along the (101) surface (Figure 1b) and investigated its band structure and optical properties. As we will discuss later in the experimental structural characterizations of our SnO2 samples, (101) and the equivalent surfaces are among the most stable terminations, thus the calculation results here are representative. As shown in Figure 1c, after the truncation, the fundamental gap becomes dipole transition allowed, that is, the band-edge emission is recovered, and the bandgap is reduced due to the formation of the surface-dangling bond state. The corresponding partial charge density mapping of valence-band maximum and conduction-band minimum (Figure 1b) confirms that the surface regions and the associated local symmetry breaking are responsible for the interband transition and bandgap reduction. We find that the physics behind this phenomenon is general and applicable to other surfaces and even other oxides.

Conceptual illustration of breaking of the dipole-forbidden transition rule in SnO2 surfaces and nanocrystals. (a) Band structure and the corresponding optical absorption spectra of bulk SnO2 calculated using generalized gradient approximation. (b) Crystal structure (left), partial charge density mappings of valence-band maximum (VBM) (middle) and conduction-band minimum (CBM) (right) of the SnO2 (101) surface. (c) Band structure and the corresponding optical absorption spectrum of the SnO2 (101) surface. (d) Partial charge densities of lowest unoccupied molecular orbital (left) and highest occupied molecular orbital (right) of a quantum dot (QD) with diameter of 1.5 nm. (e) Energy levels and the corresponding optical absorption spectrum of SnO2 QD. For the ideal bulk SnO2 with infinite dimensions, the absence of optical transition between VBM and CBM (corresponding to the fundamental bandgap) indicates that the band-edge light emission is dipole forbidden as a result of the symmetry of the band-edge wavefunctions. For the (101) surface and the QD with defects, the fundamental bandgap becomes dipole transition allowed, so the optical gap is largely reduced. As a consequence, the dipole-forbidden transition rule breaks down in SnO2 surfaces and QDs.
Nanocrystals are characterized by large surface-to-volume ratios, so they could be more effective to recover the band-edge transition in dipole-forbidden materials. To test this hypothesis, we theoretically investigated the properties of SnO2 quantum dots (QDs) with a diameter of ∼1.5 nm (Figure 1d). Several structural models were constructed, and both pseudo-hydrogen and real hydrogen were used to passivate the dangling bonds on the QD surface. We found that, if the QD is built with a high structural perfection and symmetry, the optical transition between the highest occupied molecular orbital and the lowest unoccupied molecular orbital is forbidden, resembling the bulk property. However, the transition is allowed if the QD surface is reconstructed with low symmetry (Figure 1e), indicating that a certain degree of surface structural imperfection is needed to recover the band-edge light emission in SnO2 nanocrystals. In this case, owing to the different orientations of the QDs, the optical polarization effect is not significant.
Considering the fact that oxygen vacancy (VO) is often the most stable defect in wide-bandgap oxides including SnO2, we also built a model of QDs with an VO inside to see the possible effect of defects on optical properties. The result is shown in the Supplementary information (Supplementary Figure S1). We found that the VO inside the QD reduces the optical bandgap as large as ∼0.6 eV by GGA, thus it may be responsible for the visible light absorption/emission, which is often observed in SnO2 samples. As the band-edge UV emission is the focus of this work, we did not carry out detailed studies on the effects of defect types and concentrations. Nevertheless, detailed elucidation on the roles of defects in light emission of SnO2-related materials will shed light on the structure–property correlation, and it clearly warrants further investigations.
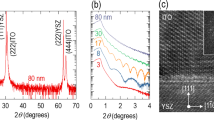
Experimentally, to realize and tune the band-edge UV emission in nanostructure-engineered SnO2 with high surface-to-volume ratios, we produced uniform SnO2 nanocrystals in the amorphous thin film matrix via controlled annealing and crystallization. Figure 2a shows the X-ray diffraction patterns of the as-grown, 400, 600 and 800 °C annealed SnO2 thin films grown on quartz substrates. For the as-grown amorphous SnO2 film, no diffraction peak was observed. After annealing at 400 °C for 30 min, the X-ray diffraction pattern exhibits three weak diffraction peaks corresponding to the (110), (101) and (211) planes, indicating that crystallized grains of rutile SnO2 emerge in the amorphous matrix. These diffraction peaks intensify and become sharper as the annealing temperature increases, and the progressive growth of crystalline grains also leads to rougher surfaces (Supplementary Figure S2 in Supplementary information).

Structural characterizations of nanostructure-engineered SnO2 thin films. (a) X-ray diffraction patterns of as-grown, 400, 600 and 800 °C annealed SnO2 thin films. The appearance of (110), (101) and (211) diffraction peaks after annealing indicates that the as-grown amorphous SnO2 thin films have been gradually crystallized. The reference powder diffraction data is also presented. (b) Raman spectra of the as-grown and the annealed SnO2 thin films. The A1g mode is enhanced and shifts to higher wave numbers with the increasing annealing temperature, suggesting enhanced crystallization. (c) High-resolution transmission electron microscope image of the SnO2 film annealed at 400 °C, showing nanocrystals embedded in the amorphous SnO2 matrix. (d) Magnified image of the nanocrystal marked in (c). (e) Corresponding selected area electron diffraction pattern taken on the 400 °C annealed sample.
To further investigate the crystalline structure and the evolution of grain size, Raman spectroscopy measurements were performed. As shown in Figure 2b, for the as-grown SnO2 film, there is no obvious Raman mode, consistent with its amorphous nature.37 After annealing, two Raman peaks were observed, and their intensities were significantly enhanced with increasing annealing temperature. More importantly, for these three annealed samples, the mode A1g peaks were found at 629.9, 634.3 and 635.7 cm−1, respectively. These wave numbers are systematically lower than the bulk SnO2 value of 638 cm−1. This reconciles with the previous observation that the A1g mode is sensitive to the grain size and shifts to lower wave numbers with decreasing grain sizes.38 From this relationship and the experimentally observed peak shift, the average grain size of SnO2 film can be evaluated to be about 3, 10 and 20 nm for the samples annealed at 400, 600 and 800 °C, respectively.
We also directly examined the annealing-induced structural evolution in the SnO2 thin films using transmission electron microscopy. Figure 2c shows the high-resolution transmission electron microscope image of the 400 °C annealed SnO2 film. It was clearly observed that nanocrystals are surrounded by the amorphous matrix, forming a nanocrystalline/amorphous hybrid structure. The distribution of the nanocrystal size is in the range of 2–6 nm with a mean size of 3.7 nm (Supplementary Figure S3, Supplementary information). Figure 2d shows the magnified image of a nanocrystal, and the lattice spacing of 0.265 nm reconciles with the (101) planes of rutile SnO2. Figure 2e shows the corresponding selected area electron diffraction pattern; a number of spots are superimposed on the rutile SnO2 diffraction rings, suggesting the presence of randomly oriented nanocrystals.
The annealing treatments and tailored crystallization give rise to modified optical properties in the SnO2 thin films. Figure 3a presents the optical absorption spectra of the annealed samples. For comparison, the optical absorption of a fully crystallized bulk-like SnO2 thin film grown directly at 600 °C was also measured. A strong band tail was observed in the 400 °C annealed sample, which may have a defect-related origin. As the annealing temperature increases, the absorption edges of the samples show significant blueshifts and approach the bandgap of bulk SnO2.

Optical properties of nanostructure-engineered SnO2 thin films. (a) Room temperature (RT) optical absorption spectra of the SnO2 thin films. A strong absorption tail was observed in the 400 °C annealed sample. With increasing annealing temperatures, the absorption edges significantly blueshift and approach the bandgap of bulk SnO2 as a result of enhanced crystallization. As a reference sample, the bulk-like crystalline SnO2 film was deposited directly at 600 °C. (b) RT photoluminescence (PL) spectra of the SnO2 thin films. The ultraviolet (UV) emission is much sharper in the 400 °C annealed samples compared with the others. As the annealing temperature increases, the UV emission becomes weaker, and eventually it is replaced by a broad band in the visible region, which is characteristic of the bulk SnO2. The inset shows the PL spectrum with fine structures of the 400 °C annealed sample measured at 10 K.
Importantly, this strong structure–property correlation is also reflected in the luminescence properties of the SnO2 thin films. As shown in Figure 3b, a sharp UV light emission peak emerges at 378 nm in the 400 °C annealed sample, and it becomes much broader and extends into the blue-violet region after annealing at 600 °C. It should be noted that the visible emission is much suppressed in these low-temperature annealed samples, indicating a reduced density of deep-level defects.18 However, the visible emission dominates in the 800 °C annealed sample, similar to the spectrum of the crystalline thin films. These systematic results indicate that the low-temperature (<600 °C) annealing is effective to realize the UV emission in nanocrystal-containing SnO2 films while suppressing the visible emission. Although weak band-edge emission could still be detected in other samples, enhanced UV light emission was only observed in the 400 °C annealed sample, which is featured with a high surface-to-volume ratio.
To shed light on the origin of the UV emission, the PL spectrum of the 400 °C annealed sample was collected at 10 K. As shown in the inset of Figure 3b, some fine structures were observed in the UV region, including four peaks located at 369.1 nm (3.360 eV), 374.9 nm (3.308 eV), 383.2 nm (3.236 eV) and 392.0 nm (3.163 eV). More details can be found in Supplementary Figures S4, S5 and S6 and Supplementary information. The origin of the peak at 369.1 nm can be assigned to a combination of radiative recombination of free-exciton, conduction band to acceptors (eA0) and neutral-donor-bound excitons (D0X).28, 31, 39 The peak at 374.9 nm can be attributed to the transition of donor–acceptor pairs (DAP), which is in good agreement with the results on SnO2 nanowires.30, 31 The other two weaker bands are separated from the DAP peak by ∼72 and ∼145 meV, respectively, which coincides with the phonon energy of the A1g Raman mode of the rutile SnO2 (∼77 meV). Therefore, they can be ascribed to the first- and the second-order longitudinal optical (LO) phonon replica of DAP, that is, DAP-1LO and DAP-2LO.31 It is clear that further in-depth investigations are needed to correlate the optical properties with the electronic band structure of SnO2.
One salient feature of our hybrid thin films is that a large amount of dangling bonds exists at the surface of the nanocrystalline grains, which are buried in the surrounding amorphous matrix. For ionic oxides, the dangling bonds induce shallow states,40 and the associated bound excitons are responsible for the observed UV emission. The observed UV emission unambiguously suggests that the quantum mechanical dipole-forbidden transition rule breaks down in the nanocrystalline/amorphous hybrid SnO2 thin films. It is noteworthy that the nanocrystals experimentally realized here are much larger than the simulated QDs shown in Figure 1, thus the quantum confinement effect is not significant. Furthermore, the band structure of nanostructured ionic semiconductors is sensitive to how the surfaces are passivated, and the effective bandgap can be reduced when the nanocrystal surface has dangling bonds as a result of incomplete passivation.41, 42 This appears to be the cause of the redshift of the PL peak in the 400 °C annealed sample relative to the bandgap of bulk SnO2.
An important milestone toward applying these hybrid nanocrystalline/amorphous SnO2 thin films in optoelectronics would be the observation of EL. To our knowledge, EL in the UV regime has not yet been reported in SnO2 films. For this purpose, we fabricated SnO2/p-GaN and SnO2/MgO/p-GaN heterojunction devices on p-GaN/sapphire substrates based on the 400 °C annealed SnO2 thin films. The detailed structure of the EL devices can be found in Supplementary Figure S7 and Supplementary information.
Figure 4a shows the current-voltage characteristics of the SnO2/p-GaN and the SnO2/MgO/p-GaN heterojunctions, and good rectification was obtained in both junctions. The turn-on voltages are 6 and 9 V for the SnO2/p-GaN and SnO2/MgO/p-GaN heterojunctions, respectively. To confirm that the rectification is intrinsic, we verified that the Ni/Au contacts to the p-GaN layer and the ITO/In contacts to the n-SnO2 layer are ohmic (inset of Figure 4a). As shown in Figure 4b, the room temperature PL spectra of both junctions exhibit a broad band located at ∼445 nm, which originates from the GaN layer. A SnO2-related peak at ∼378 nm was clearly observed in the PL spectrum of the SnO2/MgO/p-GaN heterojunction, indicating that the thin dielectric MgO layer efficiently blocks the leakage of excited carriers and preserves the SnO2 emission.

Performance of SnO2-based heterojunction light-emitting diodes (LEDs). (a) I–V curves of the SnO2/p-GaN and the SnO2/MgO/p-GaN heterojunctions. The turn-on voltages are about 6 and 9 V for SnO2/p-GaN and SnO2/MgO/p-GaN, respectively. Inset in (a) shows the ohmic behavior of the n-n and the p-p contacts in the LED device. (b) Normalized room temperature (RT) photoluminescence spectra of the p-GaN substrate, the SnO2/p-GaN and the SnO2/MgO/p-GaN heterojunctions. (c and d) RT electroluminescence (EL) spectra of the SnO2/p-GaN and the SnO2/MgO/p-GaN heterojunction LEDs under various forward excitation currents. Inset in (d) shows the photograph of the SnO2/MgO/p-GaN LEDs biased under a forward current of 6 mA. (e and f) Schematics illustrating the band alignment of the SnO2/p-GaN and the SnO2/MgO/p-GaN heterojunctions under a forward bias. In the SnO2/p-GaN junction, the charge leakage makes the GaN-related emission dominate the spectra. On the other hand, in the SnO2/MgO/p-GaN junction, electrons in the SnO2 layer are blocked by the high barrier at the SnO2/MgO interface, giving rise to the efficient SnO2-related EL in the ultraviolet regime.
Figures 4c and d shows the room temperature EL spectra of the SnO2/p-GaN and SnO2/MgO/p-GaN heterojunction LEDs under various forward excitation currents. In both junctions, the EL intensity increases with the excitation current. The EL spectra of the SnO2/p-GaN heterojunction exhibit a broad luminescence band spanning across both UV and blue regions, and the main GaN-related peak is centered at ∼445 nm. In contrast, the EL spectra of the SnO2/MgO/p-GaN heterojunction are featured by a much narrower emission peak located at ∼398 nm with full width at half maximum of ∼53 nm. Thus, the application of the MgO dielectric layer with a thickness of ∼15 nm successfully suppresses the 445-nm blue emission from GaN while enhancing the UV emission from SnO2. The emission spectra are inhomogeneously broadened due to the distribution of light-emitting nanocrystal sizes. The inset of Figure 4d shows the photograph of the SnO2/MgO/p-GaN LED under the forward bias current of 6 mA, where many bright emission spots were clearly observed in the junction region. This is the first reported observation of EL in SnO2, and further optimization of the synthesis and device in the future is expected to boost the lighting efficiency.43
To further shed light on the origin of the EL emissions, the schematic diagrams of band alignments in the SnO2/p-GaN and the SnO2/MgO/p-GaN heterojunctions under a forward bias are illustrated in Figures 4e and f, respectively. Considering that the electron affinities of SnO2, MgO and GaN are 4.53, 0.80 and 4.20 eV, respectively,44, 45, 46 and considering their individual bandgaps, we can deduce a conduction-band offset (CBO) of −0.33 eV and a valence-band offset (VBO) of −0.53 eV at the SnO2/p-GaN interface. Similarly, for the SnO2/MgO/p-GaN heterojunction, the CBO is −3.73 eV at the SnO2/MgO interface, and the VBO is −0.9 eV at the MgO/p-GaN interface. For the SnO2/p-GaN heterojunction, electrons in the SnO2 layer can leak into the p-type GaN layer and recombine radiatively with holes because of the small CBO at the interface, and vice versa for holes in the GaN layer. As a result, the emission band at 445 nm from the GaN layer dominates the EL spectra of the SnO2/p-GaN junction, whereas the SnO2-related emission at the shorter wavelength is almost invisible. In contrast, the band alignment and carrier recombination are drastically modified when a MgO dielectric layer is applied in the heterojunction, where electrons are effectively confined within the SnO2 layer owing to the large CBO (−3.73 eV) and the band bending at the SnO2/MgO interface. On the other hand, holes in the p-GaN layer can still be injected into the SnO2 layer because of the small VBO (−0.9 eV) at the MgO/p-GaN interface. Consequently, efficient radiative recombinations of carriers happen at the SnO2/MgO interface, giving out the observed bright UV light. Meanwhile, the GaN-related emission band at the longer wavelength is effectively suppressed in the SnO2/MgO/p-GaN heterojunction. Thus, the MgO dielectric layer is the key factor in effectively improving the performance of the SnO2-based LEDs.
In summary, our results suggest that tailoring the nanocrystalline structure of wide-bandgap oxides and engineering the symmetry of surface states is an effective route to achieving novel optical properties that are absent or weak in the bulk counterparts. The observation of UV band-edge emission is a signature of the alteration of the band symmetry and the breakdown of the quantum mechanical dipole-forbidden transition rule. Furthermore, using this approach, we fabricated the first UV LED heterostructure based on nanostructured SnO2 thin films, which may find extensive applications in optoelectronics, displays and solid-state lighting. Our methodology is transferable to other semiconductors with ‘forbidden’ energy gaps, such as In2O3, TiO2, Cu2O, Ag2O and GeO2, offering a promising route toward adding new members to the family of light-emitting materials.
References
Look, D. C., Hemsky, J. W. & Sizelove, J. R. Residual native shallow donor in ZnO. Phys. Rev. Lett. 82, 2552–2555 (1999).
Walsh, A., Da Silva, J. L. F., Wei, S.-H., Körber, C., Klein, A., Piper, L. F. J., DeMasi, A., Smith, K. E., Panaccione, G., Torelli, P., Payne, D. J., Bourlange, A. & Egdell, R. G. Nature of the band gap of In2O3 revealed by first-principles calculations and X-ray spectroscopy. Phys. Rev. Lett. 100, 167402 (2008).
Li, Y. F., Deng, R., Yao, B., Xing, G. Z., Wang, D. D. & Wu, T. Tuning ferromagnetism in MgxZn1-xO thin films by band gap and defect engineering. Appl. Phys. Lett. 97, 102506 (2010).
Jain, M., Chelikowsky, J. R. & Louie, S. G. Quasiparticle excitations and charge transition levels of oxygen vacancies in Hafnia. Phys. Rev. Lett. 107, 216803 (2011).
Peng, H. Y., Li, Y. F., Lin, W. N., Wang, Y. Z., Gao, X. Y. & Wu, T. Deterministic conversion between memory and threshold resistive switching via tuning the strong electron correlation. Sci. Rep. 2, 442 (2012).
Batzill, M. & Diebold, U. The surface and materials science of tin oxide. Prog. Surf. Sci. 79, 47–154 (2005).
Vinodgopal, K., Bedja, I. & Kamat, P. V. Nanostructured semiconductor films for photocatalysis. Photoelectrochemical behavior of SnO2/TiO2 composite systems and its role in photocatalytic degradation of a textile Azo dye. Chem. Mater. 8, 2180–2187 (1996).
Harrison, P. G. & Willett, M. J. The mechanism of operation of tin(iv) oxide carbon monoxide sensors. Nature 332, 337–339 (1988).
Kowal, A., Li, M., Shao, M., Sasaki, K., Vukmirovic, M. B., Zhang, J., Marinkovic, N. S., Liu, P., Frenkel, A. I. & Adzic, R. R. Ternary Pt/Rh/SnO2 electrocatalysts for oxidizing ethanol to CO2 . Nat. Mater. 8, 325–330 (2009).
Zhang, Z., Gao, J., Wong, L. M., Tao, J. G., Liao, L., Zheng, Z., Xing, G. Z., Peng, H. Y., Yu, T., Shen, Z. X., Huan, C. H. A., Wang, S. J. & Wu, T. Morphology-controlled synthesis and a comparative study of the physical properties of SnO2 nanostructures: from ultrathin nanowires to ultrawide nanobelts. Nanotechnology 20, 135605 (2009).
Qian, J., Liu, P., Xiao, Y., Jiang, Y., Cao, Y., Ai, X. & Yang, H. TiO2-coated multilayered SnO2 hollow microspheres for dye-sensitized solar cells. Adv. Mater. 21, 3663–3667 (2009).
Snaith, H. J. & Ducati, C. SnO2-based dye-sensitized hybrid solar cells exhibiting near unity absorbed photon-to-electron conversion efficiency. Nano Lett. 10, 1259–1265 (2010).
Li, Y., Deng, R., Tian, Y., Yao, B. & Wu, T. Role of donor-acceptor complexes and impurity band in stabilizing ferromagnetic order in Cu-doped SnO2 thin films. Appl. Phys. Lett. 100, 172402 (2012).
Yu, B., Zhu, C. & Gan, F. Exciton spectra of SnO2 nanocrystals with surficial dipole layer. Opt. Mater. 7, 15–20 (1997).
Agekyan, V. T. Spectroscopic properties of semiconductor crystals with direct forbidden energy gap. Phys. Status Solidi A 43, 11–42 (1977).
Fröhlich, D., Kenklies, R. & Helbig, R. Band-gap assignment in SnO2 by two-photon spectroscopy. Phys. Rev. Lett. 41, 1750–1751 (1978).
Hu, J. Q., Ma, X. L., Shang, N. G., Xie, Z. Y., Wong, N. B., Lee, C. S. & Lee, S. T. Large-scale rapid oxidation synthesis of SnO2 nanoribbons. J. Phys. Chem. B 106, 3823–3826 (2002).
Hu, J., Bando, Y., Liu, Q. & Golberg, D. Laser-ablation growth and optical properties of wide and long single-crystal SnO2 ribbons. Adv. Func. Mater. 13, 493–496 (2003).
Faglia, G., Baratto, C., Sberveglieri, G., Zha, M. & Zappettini, A. Adsorption effects of NO2 at ppm level on visible photoluminescence response of SnO2 nanobelts. Appl. Phys. Lett. 86, 011923 (2005).
He, J. H., Wu, T., Hsin, C., Li, K., Chen, L., Chueh, Y., Chou, L. & Wang, Z. Beaklike SnO2 nanorods with strong photoluminescent and field-emission properties. Small 2, 116–120 (2006).
Luo, S., Fan, J., Liu, W., Zhang, M., Song, Z., Lin, C., Wu, X. & Chu, P. K. Synthesis and low-temperature photoluminescence properties of SnO2 nanowires and nanobelts. Nanotechnology 17, 1695–1699 (2006).
Zhou, J. X., Zhang, M. S., Hong, J. M. & Yin, Z. Raman spectroscopic and photoluminescence study of single-crystalline SnO2 nanowires. Solid State Commun. 138, 242–246 (2006).
Luo, S., Chu, P. K., Liu, W., Zhang, M. & Lin, C. Origin of low-temperature photoluminescence from SnO2 nanowires fabricated by thermal evaporation and annealed in different ambients. Appl. Phys. Lett. 88, 183112 (2006).
Lettieri, S., Causà, M., Setaro, A., Trani, F., Barone, V., Ninno, D. & Maddalena, P. Direct role of surface oxygen vacancies in visible light emission of tin dioxide nanowires. J. Chem. Phys. 129, 244710 (2008).
Lettieri, S., Setaro, A., Baratto, C., Comini, E., Faglia, G., Sberveglieri, G. & Maddalena, P. On the mechanism of photoluminescence quenching in tin dioxide nanowires by NO2 adsorption. New J. Phys. 10, 043013 (2008).
Blattner, G., Klingshirn, C. & Helbig, R. Impurity transitions in the photoluminescence spectra of SnO2 . Solid State Commun. 33, 341–344 (1980).
Jeong, J., Choi, S.-P., Chang, C. I., Shin, D. C., Park, J. S., Lee, B. T., Park, Y.-J. & Song, H.-J. Photoluminescence properties of SnO2 thin films grown by thermal CVD. Solid State Commun. 127, 595–597 (2003).
Chen, R., Xing, G. Z., Gao, J., Zhang, Z., Wu, T. & Sun, H. D. Characteristics of ultraviolet photoluminescence from high quality tin oxide nanowires. Appl. Phys. Lett. 95, 061908 (2009).
Kar, A., Stroscio, M. A., Dutta, M., Kumari, J. & Meyyappan, M. Observation of ultraviolet emission and effect of surface states on the luminescence from tin oxide nanowires. Appl. Phys. Lett. 94, 101905 (2009).
Yang, H. Y., Yu, S. F., Lau, S. P., Tsang, S. H., Xing, G. Z. & Wu, T. Ultraviolet coherent random lasing in randomly assembled SnO2 nanowires. Appl. Phys. Lett. 94, 241121 (2009).
Yang, H. Y., Yu, S. F., Tsang, S. H., Chen, T. P., Gao, J. & Wu, T. High temperature excitonic lasing characteristics of randomly assembled SnO2 nanowires. Appl. Phys. Lett. 95, 131106 (2009).
Yuan, Z. Z., Li, D. S., Wang, M. H., Chen, P. L., Gong, D. R., Cheng, P. H. & Yang, D. R. Electroluminescence of SnO2/p-Si heterojunction. Appl. Phys. Lett. 92, 121908 (2008).
Wang, Y., Ma, J., Ji, F., Yu, X. & Ma, H. Structural and photoluminescence characters of SnO2:Sb films deposited by RF magnetron sputtering. J. Lumin. 114, 71–76 (2005).
Kresse, G. & Furthmüller, J. Efficient iterative schemes for ab initio total-energy calculations using a plane-wave basis set. Phys. Rev. B 54, 11169–11186 (1996).
Blöchl, P. E. Projector augmented-wave method. Phys. Rev. B 50, 17953–17979 (1994).
Adolph, B., Furthmüller, J. & Bechstedt, F. Optical properties of semiconductors using projector-augmented waves. Phys. Rev. B 63, 125108 (2001).
Ristic, M., Ivanda, M., Popovic, S. & Music, S. Dependence of nanocrystalline SnO2 particle size on synthesis route. J. Non-Cryst. Solids 303, 270–280 (2002).
Dieguez, A., Romano-Rodriguez, A., Vila, A. & Morante, J. R. The complete Raman spectrum of nanometric SnO2 particles. J. Appl. Phys. 90, 1550–1557 (2001).
Liu, B., Cheng, C. W., Chen, R., Shen, Z. X., Fan, H. J. & Sun, H. D. Fine structure of ultraviolet photoluminescence of tin oxide nanowires. J. Phys. Chem. C 114, 3407–3410 (2010).
Walsh, A., Da Silva, J. L. F. & Wei, S.-H. Interplay between order and disorder in the high performance of amorphous transparent conducting oxides. Chem. Mater. 21, 5119–5124 (2009).
Deng, H.-X., Li, S.-S., Li, J. & Wei, S.-H. Effect of hydrogen passivation on the electronic structure of ionic semiconductor nanostructures. Phys. Rev. B 85, 195382 (2012).
Chen, X., Liu, L., Yu, P. Y. & Mao, S. S. Increasing solar absorption for photocatalysis with black hydrogenated titanium dioxide nanocrystals. Science 331, 746–750 (2011).
Pimputkar, S., Speck, J. S., DenBaars, S. P. & Nakamura, S. Prospects for LED lighting. Nat. Photon. 3, 180–182 (2009).
Islam, M. N. & Hakim, M. O. Electron affinity and work function of polycrystalline SnO2 thin film. J. Mater. Sci. Lett. 5, 63–65 (1986).
Qiao, D., Yu, L. S., Lau, S. S., Redwing, J. M., Lin, J. Y. & Jiang, H. X. Dependence of Ni/AlGaN Schottky barrier height on Al mole fraction. J. Appl. Phys. 87, 801–804 (2000).
Thomas, R. E., Gibson, J. W. & Haas, G. A. Low energy electron reflection (LEER) and electron affinity of MgO. Appl. Surf. Sci. 5, 398–405 (1980).
Acknowledgements
We acknowledge support from the National Research Foundation of Singapore. The work at NREL was funded by the US Department of Energy under the Grant No. DE-AC36-08GO28308.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Supplementary Information accompanies the paper on the NPG Asia Materials website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Li, Y., Yin, W., Deng, R. et al. Realizing a SnO2-based ultraviolet light-emitting diode via breaking the dipole-forbidden rule. NPG Asia Mater 4, e30 (2012). https://doi.org/10.1038/am.2012.56
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/am.2012.56
Keywords
This article is cited by
-
Photocatalytic Degradation of Crystal Violet Using SnO2/ZnO Nanocomposite Synthesized by Facile Sol-Gel Method
Journal of Cluster Science (2024)
-
Green Synthesis of Stannic Oxide Nanoparticles for Ciprofloxacin Degradation: Optimization and Modelling Using a Response Surface Methodology (RSM) Based on the Box–Behnken Design
Journal of Cluster Science (2023)
-
High-spatial-resolution composition analysis of micro/nano-structures with a nanoscale compositional variation
Nano Research (2023)
-
Exploring dopant effects in stannic oxide nanoparticles for CO2 electro-reduction to formate
Nature Communications (2022)
-
Structural, morphological, optical and electrical properties of Ni-doped SnO2 thin films by pneumatic spray pyrolysis method
Bulletin of Materials Science (2022)
