
ABSTRACT
Seven different, but highly conserved 14-3-3 proteins are involved in diverse signaling pathways in human cells. It is unclear how the 14-3-3σ isoform, a transcriptional target of p53, exerts its inhibitory effect on the cell cycle in the presence of other 14-3-3 isoforms, which are constitutively expressed at high levels. In order to identify structural differences between the 14-3-3 isoforms, we solved the crystal structure of the human 14-3-3σ protein at a resolution of 2.8 Å and compared it to the known structures of 14-3-3ζ and 14-3-3τ. The global architecture of the 14-3-3σ fold is similar to the previously determined structures of 14-3-3ζ and 14-3-3τ: two 14-3-3σ molecules form a cup-shaped dimer. Significant differences between these 14-3-3 isoforms were detected adjacent to the amphipathic groove, which mediates the binding to phosphorylated consensus motifs in 14-3-3-ligands. Another specificity determining region is localized between amino-acids 203 to 215. These differences presumably select for the interaction with specific ligands, which may explain the different biological functions of the respective 14-3-3 isoforms. Furthermore, the two 14-3-3σ molecules forming a dimer differ by the spatial position of the ninth helix, which is shifted to the inside of the ligand interaction surface, thus indicating adaptability of this part of the molecule. In addition, 5 non-conserved residues are located at the interface between two 14-3-3σ proteins forming a dimer and represent candidate determinants of homo- and hetero-dimerization specificity. The structural differences among the 14-3-3 isoforms described here presumably contribute to isoform-specific interactions and functions.
Similar content being viewed by others


INTRODUCTION
Of the seven human 14-3-3 isoforms (designated as β, ε, γ, η, σ, τ, ζ), the 14-3-3σ protein has been linked to cancer most directly (for reviews see 1, 2). The expression of 14-3-3σ is restricted to epithelial cells. In human keratinocytes 14-3-3σ expression is induced during differentiation and is necessary for the exit from the stem cell compartment, whereas its loss allows bypass of senescence 3. 14-3-3σ expression is lost in numerous carcinomas either due to epigenetic silencing by CpG-methylation, which has been detected in a large number of different tumor types 1, 2, or due to mutation of p53, which directly induces expression of 14-3-3σ 4. Epigenetic silencing of 14-3-3σ has been detected at a high frequency in carcinomas of the breast 5, ovary 6, endometrium 6, prostate 7, skin 8, lung 9 and liver 10. After DNA-damage, p53-induced 14-3-3σ expression mediates a cell cycle arrest in the G2-phase 4, presumably by cytoplasmic sequestration of CDC2/cyclin B1 complexes 11. Experimental removal of the 14-3-3σ gene prevents a stable G2/M-arrest after DNA-damage and sensitizes cells to DNA-damaging treatments commonly used in cancer therapy 7, 11.
14-3-3 proteins form dimers that bind to protein ligands following serine/threonine-phosphorylation by basophilic kinases, such as cAMP-dependent protein kinase or PKB/AKT. Two canonical 14-3-3-binding motifs have been identified as R(S/X)XpSXP and RXXXpSXP, where pS represents phospho-serine or phospho-threonine and X any amino-acid 12. Association with 14-3-3 proteins regulates the function of ligands by inter- and intra-compartmental sequestration, activation/inactivation of enzymatic activity and promotion/inhibition of protein-interactions; thereby, numerous cellular processes in all multi-cellular species analyzed are regulated by 14-3-3 proteins (reviewed in 1, 13, 14). In part this regulation is mediated by conformational changes of the ligand after association with 14-3-3 proteins 15.
Interactions identified between other 14-3-3 isoforms and protein ligands do not necessarily apply to 14-3-3σ as distinct 14-3-3 isoforms show preferential or selective binding of ligands 16, 17, 18. Binding studies with recombinant 14-3-3 isoforms and p27 showed that 14-3-3β, ε, γ, τ and ζ isoforms, but not σ, bind to p27 and sequester p27 in the cytoplasm 17. Also, CDC25C is not bound by 14-3-3σ, however by other 14-3-3 isoforms 11. The specific elimination of certain 14-3-3 isoforms by genetic means or association of their deletion with distinct syndromes also supports the notion that isoform-specific functions and interactions exist. For example, deletion of 14-3-3ε is found in patients with Miller-Dieker Syndrome and results in a neuronal migration disorder 19. In Drosophila melanogaster 14-3-3ε is required for correct timing of mitosis in undisturbed post-blastoderm cell cycles, whereas 14-3-3ζ is required for normal chromosomal segregation during syncytial mitosis 20. In human cells the 14-3-3σ gene is the only 14-3-3 isoform induced by p53 after DNA damage and it is unknown how this induction exerts a specific effect in the presence of high levels of other 14-3-3 isoforms, which are constitutively expressed 4, 21.
So far the structures of 14-3-3τ and 14-3-3ζ have been solved 22, 23. In order to identify structural differences between the 14-3-3 isoforms, which may determine biological specificity, the crystal structure of 14-3-3σ was determined and compared to the structures of 14-3-3τ and 14-3-3ζ.
MATERIALS AND METHODS
Bacterial expression, purification and crystallization of 14-3-3σ
14-3-3σ was amplified by PCR using the primers 5′-ggtaattgagggtcgcatggagagagagccagtctgatc-3′ and 5′-agaggagagttagagcctcagctctggggctcctgggg-3′ with a previously described adenoviral expression vector as a template 4 and subcloned into pET30XaLic (Novagen). E. coli BL21 cells transformed with this plasmid were grown at 37°C to an OD600 of 0.6–0.8. To induce protein expression, IPTG was added to a final concentration of 1 mM and cells were further incubated at 28°C for 4 h. Cells were lysed in 50 mM NaH2PO4, 300 mM NaCl, 10 mM imidazole pH 8.0 and the native 6×His-tagged 14-3-3σ protein was purified using Ni-NTA-agarose (Qiagen) according to the manufacturers' protocol. For removal of the 6×His-Tag, the protein was incubated with the protease Factor Xa (Novagen) for 72 h at room temperature in a buffer containing 50 mM Tris/HCl, pH 8.0, 100 mM NaCl, and 5 mM CaCl2 (10 U Factor Xa per 1 mg protein). Further purification was performed using size exclusion chromatography with S75pg (Amersham Pharmacia) to remove Factor Xa and other minor contaminants. Initial screening for crystals was performed with Crystal Screen I and II (Hamptom Research). Crystallization of the protein was carried out with the sitting drop vapor diffusion method. The protein crystallized under many different conditions, however it was difficult to obtain well-diffracting crystals. The best crystals were obtained from 40 % PEG 4000, 0.6 M ammonium sulfate and 0.1 M Tris pH 9.0. They appeared after several weeks and grew to a final size of ca. 0.3 × 0.2 × 0.1 mm. Crystals were flash frozen after soaking for ca. 30 sec in a drop of a reservoir solution containing 30% (v/v) glycerol as a cryoprotectant. The crystals belong to the space group P212121 with unit cell dimensions a =71.65 Å, b = 80.85 Å, c = 99.13 Å and contained one dimer molecule per asymmetric unit.
Data collection and refinement
A 2.8 Å dataset was collected on the MPG/GBF beamline BW6 at DESY, Hamburg, Germany. Diffraction data for the structure refinement was collected at 90K. Diffraction images were acquired with MARCCD detectors. The summary of the data collection and refinement statistics are presented in Tab. 1. The collected data were integrated, scaled and merged by XDS and XSCALE programs 24. The structure was determined by molecular replacement using the Molrep program from the CCP4 suite 25. The structure of the 14-3-3ζ isoform taken from the PDB entry 1QJB 26 was used as a probe structure after removing the phosphopeptide part. The initial R-factor of the model was 0.46. The model was then refined by Refmac5 25 and rebuilt by XtalViev/Xfit 27. Waters were added by Arp/wArp 28. The final R crystallographic factor was 0.22 and Rfree 0.31. Most of the model had clear interpretable electron density, however, the loop regions of Glu71A-Lys77A, Ser210A-Asp212A and Glu72B-Gly73B had no interpretable density as well as certain solvent exposed side-chains, and those parts were omitted in the model (A and B denotes the two chains in the dimer, respectively; Fig. 2).

An overall ribbon plot of the 14-3-3σ homodimer. Each monomer consists of nine antiparallel helices and the protein dimerizes in a perfect 2-fold symmetry. The dimer forms a large cup-shaped space between monomers with two phosphopeptide binding sites at its sides. The C-terminal helix of the monomer A (blue) is significantly shifted toward the binding site; chain B is shown in dark-red. (A) a view perpendicular to the helices axis is presented. (B) a view parallel to this axis.
Cell culture procedures
HEK293T cells were cultured in Dulbecco's modified eagle medium (DMEM) supplemented with high glucose (Invitrogen) in 5% fetal bovine serum (Hyclone). HEK293T were transfected by calcium-phosphate precipitation.
Mammalian expression vectors
For expression in mammalian cells, the respective open reading frames were amplified by PCR using the primers 5′- acggtacccaccatggagagagccagtctg-3′ and 5′- ccggatccttgctagcgtaatctggaacatc-3′ for 14-3-3σ and 5′- agcagatctatgttccagatcccagagtttg-3′ and 5′- agcggtacctcactgggagggggcggag-3′ for BAD and inserted into pEYFP-C1 (Clontech). BAD was amplified from cDNA derived from human fibroblasts. All plasmids used were confirmed by sequence analysis.
14-3-3σ-overlay-assay
Recombinant His-tagged 14-3-3σ was expressed from pET30XaLICσ in E. coli BL21 after induction with 1 mM IPTG for 4 h at 28°C. The protein was purified using Ni-NTA-agarose (Qiagen), dialyzed to 50 mM Tris/HCl pH 8.0, 100 mM NaCl, 5 mM CaCl2, and the His-tag was removed by cleavage with factor Xa (Novagene). The recombinant 14-3-3σ protein was then labelled with digoxigenin (DIG) using the DIG-protein labelling kit (Roche) at a molar ratio of 1:70. For detection of interaction of proteins with recombinant 14-3-3σ, 50 μg of total cell lysates from transfected 293T cells were separated on 10% SDS gels and transferred to nitrocellulose membranes. The membranes were incubated with 1 μg/ml DIG-labelled-14-3-3σ in 25 mM Tris/HCl pH 7.5, 500 mM NaCl, 1 mg/ml BSA over night at 4°C. Detection of DIG-labelled-14-3-3σ was performed using anti-DIG-POD antibody (Roche). As expression control, the membranes were incubated with anti-GFP antibody (sc-8334, Santa Cruz) and anti-rabbit-HRP (Sigma). Enhanced chemoluminescence generated by secondary antibodies was detected with a CCD camera (440CF imaging system, Kodak).
Structural data deposition
Coordinates and the experimental structural factors of the 14-3-3σ dimer crystal structure are deposited in PDB under code 1YZ5.
RESULTS AND DISCUSSION
Bacterial expression of functional 14-3-3σ
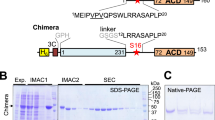
For crystallization the human 14-3-3σ protein was expressed in E. coli. Correct folding of the purified protein was confirmed by NMR (data not shown). In order to further confirm the functional integrity of recombinant 14-3-3σ protein, the direct binding of recombinant 14-3-3σ to BAD, a previously characterized interaction 29, was tested in an overlay assay (Fig. 1). Recombinant 14-3-3σ was found to interact with BAD and with 14-3-3σ expressed in HEK293T cells, indicating that the recombinant 14-3-3σ protein employed here adopts a functional conformation which allows interaction with known interacting proteins.

Binding of bacterially produced 14-3-3σ to cellular BAD protein. HEK293T cells were transiently transfected with vectors encoding for YFP-tagged BAD (BAD-YFP) or 14-3-3σ (14-3-3σ-YFP) or a control vector (YFP) and harvested 24 hours after transfection. 50 μg of total lysates were separated by SDS-PAGE, transferred to nitrocellulose membranes and incubated with DIG-labeled 14-3-3σ or anti-GFP antibodies.
Crystal structure of the 14-3-3σ dimer
The structure of the 14-3-3σ dimer was solved by molecular replacement at a resolution of 2.8 Å. A ribbon plot of the 14-3-3σ structure is shown in Fig. 2 and details of data collection and refinement statistics are presented in Tab. 1. 14-3-3σ crystallized as a homodimer. Each monomer consists of nine anti-parallel helices forming an elongated bundle. The first four helices participate in the formation of the homodimer. The remaining five helices form a deep groove providing the phosphopeptide-binding site. The overall root-mean-square-deviation (RMSD) between monomers calculated for the backbone atome is small (1.07 Å), but a clear difference is observed in the position of the ninth helix. This helix is shifted about 1.7 Å towards the ligand binding cleft in one monomer (chain A) compared to the second (chain B). As crystal packing probably causes this shift, it may indicate flexibility and adaptability of this region to structural changes that accompany ligand binding.
Structural comparison of 14-3-3σ, τ and ζ
We compared our structure of 14-3-3σ to the previously published structures of 14-3-3ζ and 14-3-3τ 12, 15, 22, 23. Only the structure of 14-3-3ζ solved by Liu et al was determined in an unliganded form, i.e. in the same state as 14-3-3σ determined here. The two additional structures of 14-3-3ζ and 14-3-3τ were determined bound to a phosphopeptide or a sulfate group, respectively. In addition, the structure of 14-3-3ζ has the same overall fold compared to the published structures of the ζ and τ isoforms15. The structure of 14-3-3σ has the same overall fold as the known fold of the ζ and τ isoforms (Fig. 3). The interface mechanism responsible for dimerization is identical for the three 14-3-3 isoforms. Most residues not conserved between isoforms are localized on the outer surface of the molecule, while the phosphopeptide-binding cleft is highly conserved (Fig. 4). The dimerization surfaces of the 14-3-3σ, ζ, and τ isoforms display several differences, which may alter the binding constant of different isoform combinations and result in limited heterodimerization among the isoforms (Figs. 4 and 8: residues in red): the most significant changes between the σ isoform and the two other isoforms are Ser5(σ)-Glu5(ζ,τ), Glu20(σ)-Asp20(ζ,τ), Phe25(σ)-Cys25(ζ,τ), Gln55(σ)-Arg55(ζ,τ) and Gln80(σ)-Met78(ζ)-Leu(τ). In the 14-3-3τ isoform the loop Glu131-Thr141 has adopted a different main chain conformation than in 14-3-3σ and ζ, however, these loop residues have the same character and it is unlikely that they affect 14-3-3 dimerization.

Comparative superposition of 14-3-3σ, ζ and τ. This main chain trace representation shows 14-3-3σ in red, 14-3-3ζ in blue and 14-3-3τ in yellow. RMSD between 14-3-3σ and 14-3-3ζ for backbone atoms are 1.15 Å, between 14-3-3σ and 14-3-3τ 1.18 Å and between 14-3-3ζ and 14-3-3τ 0.71 Å.

Comparative, structural analysis of phosphopeptide binding and dimerization interfaces in 14-3-3 proteins. Surface plot of the 14-3-3σ monomer. Residues belonging to the dimerization surface that differ significantly from other isoforms are shown in red. Residues forming the edge of the peptide binding site which differ from the 14-3-3τ and ζ isoforms are shown in dark blue. Other non-conserved residues are shown in green whereas conserved parts are in gray. The Ala203-Asp215 loop has been marked by a circle (in red line). The phosphopeptide binding region is highlighted in light green.

Alignments of primary sequences of 14-3-3σ, ζ and τ. Differences in the structures identified here are highlighted in colors. Amino acid residues shown with a black background are conserved in at least 6 of the 7 human 14-3-3 isoforms according to reference 14. The positions of α-helices (α1 to α9) are indicated by rods above the alignment. Residues in red presumably determine 14-3-3 dimerization specificity. For 14-3-3σ and 14-3-3ζ these residues are Ser5-Glu5, Glu20-Asp20, Phe25-Cys25, Gln55-Arg55 and Glu80-Met78, respectively. Residues constituting the ligand-specificity region proposed here are shown in blue for 14-3-3σ and ζ, respectively: Cys38-Asn38, Asp113-Gln111, Pro163-Gln161, Asn166-His164 and Asp211-Glu209. For comparison between 14-3-3σ and τ Asp113-Asn111 and Ala114-Pro112 are marked in blue. The yellow residues (Arg142-Gly140, Ala147-Ser145) may be of minor importance for ligand-specificity of 14-3-3σ and ζ. Three 14-3-3σ-specific residues present in the potential ligand-specificity region Ala203-Asp215 are shown in magenta (Met202, Asp204, His206).
The most significant difference between the structures of 14-3-3 isoforms solved so far and our structure of 14-3-3σ is seen in the configuration of the loop located between Ala203 and Asp215 (Fig. 5). This component of the structures is in “an open conformation” relative to the ligand-binding site in 14-3-3σ and has a different position when compared to the peptide/small molecule bound 14-3-3ζ and τ isoforms and also a different fold in the 14-3-3ζ/serotonin N-acetyltransferase complex. The movement of this fragment is large between the 14-3-3σ and the peptide bound 14-3-3ζ structures (about 4.2 Å between Cα of Ser207ζ and Ser209σ). In the structures of the 14-3-3ζ and τ isoforms this loop partly folds upon itself producing “a closed conformation”. In the structure of 14-3-3ζ bound to serotonin N-acetyltransferase, the C-terminal portion of the loop is pushed away from the phosphopeptide binding cleft by the intruding serotonin N-acetyltransferase molecule (15 PDB 1IB1). The long axis of the loop, which runs from Ala203 to Asp215, is rotated 90° relative to the position of this axis in 14-3-3σ. Taken together, the leading difference among the 14-3-3 isoforms is seen in the position of loop 203-215 relative to the phosphopeptide binding site in 14-3-3 structures. These differences suggest that this area is the major ligand-specificity region.

Ligand recognition by 14-3-3 proteins. (A) Unbound σ (red) and ζ (blue) are superimposed with the structure of the ζ isoform (green) bound to serotonin N-acetyltransferase (Cα trace) to indicate the structural diversity of the Ala203 and Asp215 loop area. (B) Structures of ζ (blue) and σ (red) are overlaid. The molecular surface of the σ isoform is shown half-transparent. The structure and surface of the phosphopeptide from the 1QJA model is shown in light blue. Non-conserved residues in this area are labeled for the σ isoform. Significant difference in the Ala203 and Asp215 loop are evident.
In the 14-3-3σ structure this loop is well determined in one monomer (chain B), and partially defined in the other (chain A, no electron density was seen for tip residues Glu210-Ser212). However, in the parts of the chain A loop with interpretable electron densities the fold of the loop is identical in both monomers, indicating that this difference is not caused by crystal packing as each monomer is in a different environment. In the only other structure of a completely free 14-3-3, the ζ isoform presented by Liu et al, this segment of the structure is missing. Our data and the structure determined by Liu et al indicate that this loop might be flexible. This feature however would not interfere with its proposed function of a major ligand-specificity region, which utilizes both sequential and structural di-versities of this region to select for isoform-specific binding partners.
The most significant divergence in amino acid types towards other 14-3-3 isoforms is seen in the same area: at the edge of the phosphopeptide-binding groove at the 14-3-3-residues marked in blue in Fig. 4. This part has a strictly preserved backbone structure among the 14-3-3 proteins and is in a significant distance (ca. 20 Å) from the primary phosphoserine binding site. Since structural properties of the binding site are analogous for the isoforms for which structures have been determined, the specific amino acids in this part are also prime candidates for determining the isoform-specificity towards ligands. The most important changes in amino acid character in this region are: Asn166(σ)-His164(ζ,τ), Pro163(σ)-Gln161(ζ,τ), Cys38(σ)-Asn38(ζ,τ), Asp209/211(σ,τ)-Glu209(ζ) and Asp113(σ)-Asn111(τ)-Gln111(ζ). In conclusion, the periphery of the phosphopeptide-binding cleft accumulates significant differences both in the fold (residues 203-215 in σ) and residue properties (the σ residues 38, 113, 163, 166, 209). Based on comparison of all mentioned structures, we propose that the Ala203 and Asp215 loop and its neighborhood represent the major ligand specificity region (Figs. 5 and 6). On the primary sequence level this loop is highly conserved between the seven isoforms, but contains 3 unique amino acids (Met202, Asp204, His206) in 14-3-3σ which are not found in any other 14-3-3 isoforms (see Fig. 8 and 14).

Proposed major ligand specificity region of 14-3-3σ. The figure shows the edge of the phosphopeptide binding groove, from the C-terminal side of a phosphopeptide. (A) the molecular surface of the 14-3-3σ isoform colored by atom charge. The structure presents an “open” conformation of the cleft edge. (B) The same area of the 14-3-3ζ isoform has a “closed” conformation. (C) Serotonin N-acetyltransferase in complex with 14-3-3ζ shown as a semi-transparent surface plot; the phosphoserine region is shown as a stick model. Except for the phosphopeptide cleft, the only possible contact area is localized at the edge of the 14-3-3ζ molecule, thus indicating that this part may be responsible for selective ligand binding.
The structure of the phosphopeptide binding site of the 14-3-3σ molecule is identical to the ζ and τ isoforms with bound ligands (structures 1QJA and 1QJB) (Fig. 7A), but not to the ζ isoform in its free form (1A4O). This difference is especially pronounced in the conformations of Trp230 and Tyr181, which form an aromatic environment at the N-terminus of a potential 14-3-3-binding peptide. As shown in Fig. 7B, the aromatic rings of Trp230 and Tyr181 in 14-3-3σ are oriented in the same plane as in the ζ isoform with a bound phosphopeptide, while in the free form of the ζ isoform they adopt a different orientation. The conformation of additional side chains of the phosphopeptide binding site in 14-3-3σ also resembles the ζ isoform complexed with a peptide more than the uncomplexed 14-3-3ζ. This conformation of 14-3-3σ could therefore result in an increased stability of 14-3-3σ-ligand interactions when compared to 14-3-3ζ interactions. As expression of 14-3-3σ is rapidly induced after DNA-damage, a displacement of other 14-3-3 isoforms or preferential binding to ligands by 14-3-3σ may therefore be possible.

Details of phosphopeptide binding pockets in the 14-3-3ζ and σ isoforms. (A) comparison of the spatial organization of phosphopeptide binding sites in different 14-3-3 structures. ζ isoforms 1QJA (dark blue; bound to a peptide) and 1A4O (gray; without a bound peptide) were superimposed onto the solved 14-3-3σ monomers (chains A and B presented in brown and green, respectively). The phosphopeptide from the 1QJA structure is shown as a bold stick model with the phosphogroup in orange. (B) the two aromatic residues that complete the binding site are shown. ζ isoforms 1QJA (dark blue) and 1A4O (gray) are compared to the σ chains A and B presented in red and green, respectively. Positions of side chains in the free σ isoform resemble those of bound ζ isoforms. The free ζ form 1A4O differs significantly from the two other models.
As discussed above, multiple examples of selective ligand binding by different 14-3-3 isoforms exist. We recently used a targeted proteomics approach to identify 14-3-3σ-associated proteins in vivo 30. Interestingly, we found that in colorectal cancer cells 14-3-3σ was only present as a homo-dimer or as a hetero-dimer with 14-3-3γ indicating selective dimerization, which may be due to the putative dimerization determinants identified here 30. The data presented here provides a basis for the experimental validation of determinants of ligand and dimerization specificity by mutational analysis.
References
Hermeking H . The 14-3-3 cancer connection. Nat Rev Cancer 2003; 3:931–43.
Lodygin D, Hermeking H . The role of epigenetic silencing of 14-3-3σ in human cancer. Cell Research 2005; 15:237–46.
Pellegrini G, Dellambra E, Golisano O, et al. p63 identifies keratinocyte stem cells. Proc Natl Acad Sci U S A 2001; 98: 3156–61.
Hermeking H, Lengauer C, Polyak K, et al. 14-3-3σ is a p53-regulated inhibitor of G2/M progression. Mol Cell 1997; 1:3–11.
Ferguson AT, Evron E, Umbricht CB, et al. High frequency of hypermethylation at the 14-3-3σ locus leads to gene silencing in breast cancer. Proc Natl Acad Sci U S A 2000; 97:6049–54.
Mhawech P, Benz A, Cerato C, et al. Downregulation of 14-3-3σ in ovary, prostate and endometrial carcinomas is associated with CpG island methylation. Mod Pathol 2005; 18:340–8
Lodygin D, Diebold J, Hermeking H . Prostate cancer is characterized by epigentic silencing of 14-3-3σ expression. Oncogene 2004; 23:9034–41.
Lodygin D, Yazdi AS, Sander CA, Herzinger T, Hermeking H . Analysis of 14-3-3σ expression in hyper-proliferative skin diseases reveals selective loss associated with CpG-methylation in basal cell carcinoma. Oncogene 2003; 22:5519–24.
Osada H, Tatematsu Y, Yatabe Y, et al. Frequent and histological type-specific inactivation of 14-3-3σ in human lung cancers. Oncogene 2002; 21:2418–24.
Iwata N, Yamamoto H, Sasaki S, et al. Frequent hypermethylation of CpG islands and loss of expression of the 14-3-3σ gene in human hepatocellular carcinoma. Oncogene 2000; 19:5298–302.
Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B . 14-3-3σ is required to prevent mitotic catastrophe after DNA damage. Nature 1999; 401:616–20.
Yaffe MB, Rittinger K, Volinia S, et al. The structural basis for 14-3-3:phosphopeptide binding specificity. Cell 1997; 91:961–71.
Aitken A, Baxter H, Dubois T, et al. Specificity of 14-3-3 isoform dimer interactions and phosphorylation. Biochem Soc Trans 2002; 30:351–60.
Yaffe MB . How do 14-3-3 proteins work?— Gatekeeper phosphorylation and the molecular anvil hypothesis. FEBS Lett 2002; 513:53–7.
Obsil T, Ghirlando R, Klein DC, Ganguly S, Dyda F . Crystal structure of the 14-3-3ζ:serotonin N-acetyltransferase complex: a role for scaffolding in enzyme regulation. Cell 2001; 105: 257–67.
Stavridi ES, Chehab NH, Malikzay A, Halazonetis TD . Substitutions that compromise the ionizing radiation-induced association of p53 with 14-3-3 proteins also compromise the ability of p53 to induce cell cycle arrest. Cancer Res 2001; 61:7030–3.
Sekimoto T, Fukumoto M, Yoneda Y . 14-3-3 suppresses the nuclear localization of threonine 157-phosphorylated p27(Kip1). Embo J 2004; 23:1934–42.
Mils V, Baldin V, Goubin F, et al. Specific interaction between 14-3-3 isoforms and the human CDC25B phosphatase. Oncogene 2000; 19:1257–65.
Toyo-oka K, Shionoya A, Gambello MJ, et al. 14-3-3ε is important for neuronal migration by binding to NUDEL: a molecular explanation for Miller-Dieker syndrome. Nat Genet 2003; 34:274–85.
Su TT, Parry DH, Donahoe B, et al. Cell cycle roles for two 14-3-3 proteins during Drosophila development. J Cell Sci 2001; 114:3445–54.
Samuel T, Weber HO, Rauch P, et al. The G2/M regulator 14-3-3σ prevents apoptosis through sequestration of Bax. J Biol Chem 2001; 276:45201–6.
Liu D, Bienkowska J, Petosa C, et al. Crystal structure of the ζ isoform of the 14-3-3 protein. Nature 1995; 376:191–4.
Xiao B, Smerdon SJ, Jones DH, et al. Structure of a 14-3-3 protein and implications for coordination of multiple signalling pathways. Nature 1995; 376:188–91.
Kabsch W . Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants. J Appl Crystallogr 1993; 26:795–800.
Collaborative Computational Project, Number 4. The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr 1994; 50:760–3.
Rittinger K, Budman J, Xu J, et al. Structural analysis of 14-3-3 phosphopeptide complexes identifies a dual role for the nuclear export signal of 14-3-3 in ligand binding. Mol Cell 1999; 4:153–66.
McRee DE . XtalView/Xfit—A versatile program for manipulating atomic coordinates and electron density. J Struct Biol 1999; 125:156–65.
Lamzin VS . Automated refinement of protein models. Acta Crystallogr D Biol Crystallogr 1993; 49:129–47.
Subramanian RR, Masters SC, Zhang H, Fu H . Functional conservation of 14-3-3 isoforms in inhibiting bad-induced apoptosis. Exp Cell Res 2001; 271:142–51.
Benzinger A, Muster N, Koch HB, Yates JR 3rd, Hermeking H . Targeted proteomic analysis of 14-3-3σ, a p53 effector commonly silenced in cancer. Mol Cell Proteomics 2005; March 18: Epub ahead of print.
Acknowledgements
We thank Steven Gamblin for providing the 14-3-3τ coordinates. Heiko Hermeking is supported by the Max-Planck-Society and the Deutsche Krebshilfe/Dr. Mildred-Scheel-Stiftung (grant 1945).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
BENZINGER, A., POPOWICZ, G., JOY, J. et al. The crystal structure of the non-liganded 14-3-3σ protein: insights into determinants of isoform specific ligand binding and dimerization. Cell Res 15, 219–227 (2005). https://doi.org/10.1038/sj.cr.7290290
Received:
Revised:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/sj.cr.7290290
Keywords
This article is cited by
-
Genome-wide identification, characterization, and expression profiling of 14-3-3 genes in legumes
Plant Biotechnology Reports (2022)
-
Hierarchized phosphotarget binding by the seven human 14-3-3 isoforms
Nature Communications (2021)
-
14-3-3ε acts as a proviral factor in highly pathogenic porcine reproductive and respiratory syndrome virus infection
Veterinary Research (2019)
-
Evaluation of 14-3-3 sigma as a potential partner of p16 in quiescence and differentiation
In Vitro Cellular & Developmental Biology - Animal (2018)
-
Expression of TaGF14b, a 14-3-3 adaptor protein gene from wheat, enhances drought and salt tolerance in transgenic tobacco
Planta (2018)
