
Abstract
The clinical, biochemical, and neuroradiologic findings and clinical follow-up of seven patients with glutaric aciduria type II are reported.
Three phenotypes of the disease are encountered: neonatal-onset form with congenital anomalies (two patients) or without congenital anomalies (three patients) and late-onset form (two patients). The neonatal-onset form presents as an overwhelming illness, with severe hypoglycemia and metabolic acidosis leading to rapid death. Frequently it is associated with perinatal energy deprivation, a neonate with low birth weight and prematurity. The late-onset form presents with intermittent episodes of vomiting, hypoglycemia, and acidosis especially after meals rich in fat and/or proteins. All parents are consanguineous and have a first- or second-degree relationship.
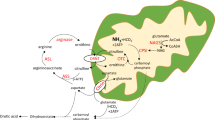
Initially, in the two phenotypes with neonatal onset and during crisis in the late-onset phenotype, routine laboratory evaluation showed severe metabolic acidosis, with an increased anion gap, hypoglycemia without ketonuria, and disturbed liver function tests. In the majority of patients with neonatal-onset forms, the kidneys, liver, and at times the spleen are enlarged with an increased echogenic pattern; however, no hepatic or renal cysts are detected. Cardiomegaly is observed in most patients. The diagnosis can be easily and rapidly reached through tandem mass spectrometry study of the blood and can further be confirmed by gas chromatography/mass spectrometry analysis of the urine organic acids.
In this report, the magnetic resonance imaging/computed tomography brain studies showed brain atrophy, white matter disease, and in one patient, fluid-filled cavities in the periventricular area and putamina. Fluorine-18-labeled 2-fluoro-2-deoxyglucose positron emission tomographic (FDG PET) brain studies in two patients with late-onset disease showed slightly decreased activity in the cerebral cortex in one and in the caudate nuclei in the other. Brain FDG PET scan and magnetic
resonance spectroscopy were normal in one patient with neonatal-onset disease.
All patients were treated with a diet low in fat and protein, oral riboflavin, and carnitine. The results were promising for the late-onset disease. Intravenous carnitine gave rewarding results in one patient with neonatal-onset disease.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout
Similar content being viewed by others

Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Al-Essa, M., Rashed, M., Bakheet, S. et al. Glutaric Aciduria Type II: Observations in Seven Patients With Neonatal- and Late-Onset Disease. J Perinatol 20, 120–128 (2000). https://doi.org/10.1038/sj.jp.7200325
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.jp.7200325
This article is cited by
-
Features and diagnostic value of body composition in patients with late-onset multiple acyl-CoA dehydrogenase deficiency
Acta Neurologica Belgica (2022)
-
Highly efficient ketone body treatment in multiple acyl-CoA dehydrogenase deficiency–related leukodystrophy
Pediatric Research (2015)
-
Riboflavin-responsive multiple acyl-CoA dehydrogenase deficiency with unknown genetic defect
Neurological Sciences (2012)
-
Cardiomyopathy in Multiple Acyl-CoA Dehydrogenase Deficiency
Pediatric Cardiology (2008)
-
FDG‐PET findings in patients with galactosaemia
Journal of Inherited Metabolic Disease (2008)
