
Abstract
Auriculo-condylar syndrome (ACS), an autosomal dominant disorder of first and second pharyngeal arches, is characterized by malformed ears (‘question mark ears’), prominent cheeks, microstomia, abnormal temporomandibular joint, and mandibular condyle hypoplasia. Penetrance seems to be complete, but there is high inter- and intra-familial phenotypic variation, with no evidence of genetic heterogeneity. We herein describe a new multigeneration family with 11 affected individuals (F1), in whom we confirm intra-familial clinical variability. Facial asymmetry, a clinical feature not highlighted in other ACS reports, was highly prevalent among the patients reported here. The gene responsible for ACS is still unknown and its identification will certainly contribute to the understanding of human craniofacial development. No chromosomal rearrangements have been associated with ACS, thus mapping and positional cloning is the best approach to identify this disease gene. To map the ACS gene, we conducted linkage analysis in two large ACS families, F1 and F2 (F2; reported elsewhere). Through segregation analysis, we first excluded three known loci associated with disorders of first and second pharyngeal arches (Treacher Collins syndrome, oculo-auriculo-vertebral spectrum, and Townes–Brocks syndrome). Next, we performed a wide genome search and we observed evidence of linkage to 1p21.1–q23.3 in F2 (LOD max 3.01 at θ=0). Interestingly, this locus was not linked to the phenotype segregating in F1. Therefore, our results led to the mapping of a first locus of ACS (ACS1) and also showed evidence for genetic heterogeneity, suggesting that there are at least two loci responsible for this phenotype.
Similar content being viewed by others


Introduction
Auriculo-condylar syndrome (ACS, OMIM 602483), an autosomal dominant disorder, was first described by Jampol et al1, who reported relatives from five generations of a family with prominent and malformed ears, microstomia, abnormal temporomandibular joint, and mandibular condyle hypoplasia. This condition is also known as question mark ear syndrome due to peculiar shape of the ear, which characteristically presents marked constriction at the junction between the lower and middle thirds of the pinna, separating the lobule from the helix and giving the ear an appearance of a ‘question mark’.2
There are only five ACS familial cases reported in the literature. Although the penetrance seems to be complete in these cases, there is high inter- and intra-familial phenotypic variation.1, 2, 3, 4 The phenotype variation is not only defined by the severity of the malformations, but also by the presence/absence of less-frequent clinical findings such as cleft palate, hypoplastic tongue, hypotonia, and developmental delay.3, 4, 5 Despite the great clinical variability, genetic heterogeneity has not been suggested for this condition. Based on the major clinical features, ACS can be classified as a disease of the first and second embryonic pharyngeal arches.
Treacher Collins syndrome (TCS; OMIM 154500), oculo-auriculo-vertebral spectrum (OAVS, OMIM 164210), and Townes–Brocks syndrome (OMIM 107480), typical disorders that involve first and second pharyngeal arch-derived structures, have several overlapping clinical signs with ACS and they should be considered for differential diagnosis. The ACS gene is still unknown and its mapping and identification will certainly bring important contributions to the understanding of the development of embryonic structures derived from these pharyngeal arches, as well as to perform differential diagnosis of the above-mentioned syndromes. As there is no chromosomal rearrangements associated to ACS, we first proposed to investigate if two large ACS families are linked to any of the loci associated with TCS (TCOF1; 5q31–32), OAVS (14q32), or Townes–Brocks syndrome (SALL1, 16q12).6 The exclusion of these three candidate regions led us to perform a wide genome search. One of the families is herein described for the first time, while the other one had already been reported.2
Subject and methods
Family data
Family 1 (F1)
The family was seen by clinicians from the Assistance Center for Cleft Lip and Palate (CAIF), Curitiba, Paraná, Brazil. Clinical evaluation included general examination with particular concern to the face and mouth, and complementary evaluation included skull and facial X-rays, whenever possible. Audiological evaluation included puretone audiometry, speech audiometry, tympanometry, stapedius reflex measurement, and brainstem-evoked response.
Family 2 (F2)
This family was originally reported by Guion-Almeida et al.2 Since its description, no additional affected child was born in the family.
DNA analysis
DNA was extracted according to standard techniques.7, 8 The study protocols were previously approved by the ethical committee of our institution and informed consent was obtained from patients or from their legal tutors.
Linkage analysis
Segregation analysis was performed using highly informative dinucleotide repeats microsatellite markers, with an average inter-marker distance of 10 cM across the genome, from ABI PRISM® Linkage Mapping Set version 2.0 (Perkin-Elmer, Applied Biosystems). The PCR products amplified by fluorescently-labeled primers were loaded onto the automated sequencer Megabace 1000 (Amersham, GE Healthcare) according to manufacturer's protocol and analyzed with Megabace Genetic Profiler® software (Amersham, GE Healthcare). We first analyzed three known candidate regions (5q31–32, 14q32, 16q12.1) for craniofacial malformations in both families using 16 markers. We next performed genome-wide scan with 370 markers spaced in average 10 cM. Linkage analysis was initially performed only in affected individuals from F2. Once we detected a shared chromosomal region among the affected patients, we extended the analysis for all the members of the F2 family using the markers from these candidate chromosomal regions. F1 was tested only for those chromosome markers that remained with positive LOD scores after this analysis due to scarcity of DNA material.
The disease was analyzed as an autosomal dominant trait, with a penetrance equal to 100% and disease gene frequency of 0.001. Two-point LOD scores were calculated using the MLINK program from FASTLINK package version 5.1 (http://linkage.rockfeller.edu/soft/). Recombination frequencies in the two-point analysis were assumed to be equal in male and female members and allele frequencies were considered to be 1/N (N=number of different alleles observed on the pedigree). We also screened 20 control individuals to calculate the allele frequencies for the markers D1S2726, D1S252, D1S498, and D1S484, which showed evidence of linkage with the disease. These allelic frequencies were used for two-point final LOD score calculations. For multipoint LOD score, we used Merlin 1.0.1 software and allelic frequencies were based on family data.9
Results
Clinical report of Family 1
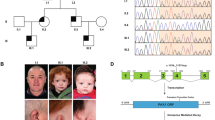
The family history revealed 11 affected individuals with ‘question mark ears’ in at least four successive generations (pedigree, Figure 1a and 2; Table 1). In addition to malformed ears, the major features of these patients were prominent cheeks, round facial appearance, micrognathia, microstomia, malocclusion, and hearing loss. After radiological examination in four individuals, temporomandibular joint abnormalities were identified, leading to the diagnosis of ACS.

Pedigrees of the studied families and haplotypes from linkage analysis in region 1p21.2–1q23.3; (a) Family 1 (F1); (b) Family 2 (F2), previously reported by Guion-Almeida et al.2

ACS-affected patients from F1. The numbers correspond to F1's genealogy number.
The penetrance seems to be complete, but there is clear clinical variable expressivity. The family comprises more affected women in the genealogy (8 affected women to 3 affected men), but no sex segregation deviation was observed according to parental transmission (P=0.33; Fisher's exact test). The most severe cases were observed in the fourth generation (individuals IV-3 and IV-6), including one (IV-7) who died in early childhood.
The ear malformations are highly variable (Figure 2), as we can observe in individual IV-3, who has auricular clefts, cupped pinna, underdevelopment and overfolding of the superior helices, pre- and post-auricular cartilaginous tags, and her mother (III-10) who has none of these features. Audiometric examinations were performed in six individuals, and all of them had at least some degree of conductive hearing loss, including individuals II-8 and III-5 who did not have stenotic ear canals. Individual II-8 had conductive and neurosensorial hearing loss of the left and right ears, respectively.
Most of the patients had malocclusion and limitations to open their mouths, but only individuals II-8, IV-6, and IV-7 had mastication and speech articulation difficulties. These three individuals also presented high-arched palate, but only IV-7 had a cleft.
We also identified facial asymmetry in a large proportion of cases, as it is evident in patients II-4, III-13, III-14, IV-3, IV-6, and IV-7 (Figure 2, Table 1). We have defined ‘facial asymmetry’ as asymmetrical (unilateral) degrees of clinical manifestation of ACS malformations, ranging from absence to presence, including the degrees of severity of the signs (from mild to severe). Intelligence was normal and no developmental delay was observed in any of the affected individuals.
Linkage study
Initially, we tested F1 and F2 for known linked loci to craniofacial anomalies (5q31–32, 14q32, and 16q12.1) but all were excluded. We thereafter conducted a genome scan in F2.
The first screening with 370 markers only in the affected individuals of F2 revealed nine regions in chromosomes 1, 2, 3, 4, 6, 15, 17, 19, and 22 with a positive LOD score (Figure 1; Supplementary Material). Of these, a maximum two-point and multipoint LOD score of 3.0 at θ=0 was observed in the analysis of the extended family only with the marker D1S252. Positive multipoint LOD scores were observed for all the markers of this region, which is from D1S206 to D1S2878 (Figure 3a and 2; Supplementary Material). The region comprised by the markers D1S206, D1S2726, D1S252, D1S498, D1S484, and D1S2878 corresponds to 1p21.1–q23.3 (about 43 cM of genetic distance) and corresponds to approximately 60 Mb of genomic sequence (Figure 4). Haplotype reconstructions lead us to conclude that the disease gene is between D1S206 (at 101.4 Mb) and D1S2878 (at 163.7 Mb). The first recombination event occurred in affected individuals II-3 and III-7, who have different alleles for D1S206. The second recombination occurred in normal individual II-9, who shares the same at-risk alleles for D1S484 and D1S2878 markers (Figure 1b).

Multipoint LOD scores for chromosome 1 markers: D1S207 (8 cM), D1S2868 (22.3 cM), D1S206 (31.6 cM), D1S2726 (40.6 cM), D1S252 (46.6 cM), D1S498 (52.6 cM), D1S484 (65.7 cM), D1S2878 (74.7 cM), and D1S196 (79.7 cM) in (a) Family 2 (F2), and (b) Family 1 (F1), both ACS-affected and normal individuals.

Mapped region 1p21.2–1q23.3 for ACS in F2. The candidate region is signaled in chromosome 1 ideogram and is expanded below. Each small square represents a reference sequence from GenBank localized in this region (approximately 250 genes). The black line signifies the region with LOD score 3.0, which includes centromeric region.
Negative LOD scores were obtained in F1 when all the available members were tested for all markers at the 1p21.1–q23.3 (multipoint LOD scores in Figure 3b; two-point for the two most informative markers, D1S252 and D1S498: Z=− ∞ at θ=0; −1.6 at θ=0.1; −0.8 at θ=0.2). These negative results were obtained because ACS-affected individuals III-10 and IV-3 do not share the same haplotype as the other affected individuals of the genealogy. In addition, individual III-8, who is normal, also has the supposed disease haplotype (Figure 1a). We also tested F1 for chromosome 3 and 19 markers that showed positive (∼1) LOD scores in F2. However, we did not observe any evidence of linkage between these markers and the disease segregating in this genealogy (Figure 3, Supplementary Material).
Discussion
The great intra- and inter-familial phenotypic variation in ACS has previously been noted and is discussed in several clinical reports. 1, 2, 4, 10 We further confirm a wide spectrum of clinical variability in the new family (F1) herein reported, in which the mildest cases presented only ‘question mark ears’, whereas the most severe ones present the full phenotype, as was the case for individual IV-7, who had several apnea episodes and died in early childhood. Besides the malformed ears, the most frequent clinical findings in F1 were round facial appearance (90%), puffy cheeks (80%), micrognathia (60%), microstomia (80%), malocclusion (88%), abnormal mandibular condyle (75%), and temporomandibular joint abnormality (100%).
F1 shares almost all of the phenotypic features described in F2, originally reported by Guion-Almeida et al2, except that none of our patients presented upslanted palpebral fissures. As we observed a high frequency of facial asymmetry in our family (60%), it would be important to evaluate if this clinical feature is present in 1p21.1–q23.3-linked ACS families. In addition to these few atypical findings, both families have the same facial ‘gestalt’ and also cleft palate and hearing loss segregating through the generations. The frequency of clinical signs from F1 was also compared with the frequency of the signs observed in previous reports (Table 2; modified from Storm et al4). Of the 16 clinical signs listed as the major ones for ACS by these authors, only seven have similar frequency in the family here described. Prominent cheeks, microstomia, hearing loss and feeding difficulties seem to be more represented in F1, but abnormal palate, macrocephaly, ptosis, mild developmental delay, microglossia, and hypotonia are in lower frequency or were not even diagnosed in F1.
Facial asymmetry is an unusual clinical signal for ACS. Surprisingly, however, it was observed in 60% of the affected individuals in F1. This is an important finding because facial asymmetry is usually considered for differential diagnosis of first and second pharyngeal arch disorders, such as OAVS, Treacher Collins, Nager (OMIM 154400), and Miller syndromes (OMIM 263750). By studying animal models of OAVS, in 1973, Poswillo12 proposed that the pathogenesis of invariably asymmetrical malformations is different from that of symmetrical malformations. This author suggested that OAVS could be explained by embryonic hemorrhage and TCS by a mechanism acting uniformly over a precise temporal period in the developing embryo, because, even inducing hemorrhage in both sides of mice embryo, symmetrical disposition of the craniofacial defects was never observed, as is the case in TCS patients. No definitive causal agent for OAVS has been discovered so far, but the gene for TCS was identified and it is known that 98% of TCS patients bearing the pathogenic mutation have bilateral and usually symmetrical malformations.13, 14, 15
We excluded three regions involved in craniofacial disorders of the first and second pharyngeal arches. This result implies that ACS is not allelic to TCS or to the known loci associated with OAVS and Townes–Brocks syndrome. It is important to mention that OAVS was linked to 14q32 region only in one familial case, and this region was excluded in other family from the same report.6 The heterogeneity of this condition has been further supported by the observation that epigenetic deregulation is associated with the occurrence of OAVS.16
Although we observed three positive regions after we have analyzed all the members of F2, an LOD score of 3.0 was only observed with markers of chromosome 1. The LOD score of 3.01 observed with the marker D1S252 is the maximum possible LOD-score value that can be obtained in F2, assuming full penetrance. These data thus suggest the evidence of linkage to the disease locus in F2 for the region 1p21.1–q23.3, and represents the first chromosomal region associated to the ACS phenotype. The candidate region (60 Mb) is very large with at least 250 genes. TBX15 (1p13; OMIM 604127), an interesting gene in this list, is expressed in the mouse embryo at day 9.5, primarily in the craniofacial region and in the developing limbs. It also belongs to the T-box gene family, composed by transcription factors involved in the regulation of developmental processes.17, 18 Another good candidate is NOTCH2 (1p13–11; OMIM 600275), which together with NOTCH1 and NOTCH3 are important genes for cell differentiation in mammals. As NOTCH2 is known to be involved in teeth development, maybe it can have other roles in craniofacial development if associated to ACS.19, 20 However, as the number of genes in this interval is very large and that there is not enough functional information of each of them, it is very difficult to select genes to be tested. Thus, it would be important to identify additional families to narrow down the mapped region and facilitate the search for the causative gene.
Interestingly, this candidate region was excluded in F1. We observed that patients III-10 and her affected daughter (IV-3) did not share any of the alleles at the putative at-risk chromosomal 1 region. These two patients present many of the similar clinical features of the other affected relatives, including facial asymmetry, and they may carry the same disease allele as the other patients in this family. Therefore, it is very likely that the condition segregating in this genealogy is caused by mutations in another locus, representing genetic heterogeneity for the ACS phenotype. We would like to propose the term ACS1 for the linked form to 1p21.1–q23.3 region, and ACS2 for the unlinked form.
The clinical data together with the linkage data reported here suggest that ACS is a very heterogeneous clinical entity, which includes many different clinical signs besides ‘question mark ears’, such as hypotonia, ptosis, microglossia, developmental delay, and facial asymmetry. Like OAVS, ACS could be classified as a ‘question mark ears’ spectrum, in which we should include ACS variations and dysgnathia complex. We also observed that the phenotype in both families became more severe through the generations; although this can be due to an ascertainment bias, it also suggests clinical anticipation. It will thus be important to evaluate this hypothesis in additional families. The identification of this disease gene will certainly contribute to our knowledge in the development of the first and second pharyngeal arches structures as well as to the understanding of OAVS conditions, as some of the isolated OAVS cases might represent subtypes of ACS.
References
Jampol M, Repetto G, Keith DA, Curtin H, Remensynder J, Holmes LB : Prominent constricted ears with malformed condyle of the mandible. Am J Med Genet 1998; 75: 449–452.
Guion-Almeida ML, Zechi-Ceide RM, Vendramini S, Kokitsu-Nakata NM : Auriculo-condilar syndrome: additional patients. Am J Med Genet 2002; 112: 209–214.
Erlich MS, Cunningham ML, Hudgins L : Transmission of the dysgnathia complex from mother to daughter. Am J Med Genet 2000; 95: 269–274.
Storm AL, Johnson JM, Lammer E, Green GE, Cunniff C : Auriculo-condylar syndrome is associated with highly variable ear and mandibular defects in multiple kindreds. Am J Med Genet 2005; 138: 141–145.
Priolo M, Lerone M, Rosaia L et al: Question mark ears temporo-mandibular joint malformation and hypotonia: auriculo-condylar syndrome or a distinct entity? Clin Dysmorph 2000; 9: 277–280.
Kelberman D, Tyson J, Chandler DC et al: Hemifacial microsomia: progress in understanding the genetic basis of a complex malformation syndrome. Hum Genet 2001; 109: 638–645.
Miller SA, Dykes DD, Polesky HF : A simple testing out procedure for extracting DNA from human nucleated cells. Nucleic Acids Res 1998; 16: 1215.
Richards B, Skoletsky J, Shuber AP et al: Multiplex PCR amplification from the CFTR gene using DNA prepared from buccal brushes/swabs. Hum Mol Genet 1993; 2: 159–163.
Abecasis GR, Cherny SS, Cookson WO, Cardon LR : Merlin-rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002; 30: 97–101.
Guion-Almeida ML, Kokitsu-Nakata NM, Zechi-Ceide RM, Vendramini S : Auriculo-condilar syndrome: further evidence for a new disorder. Am J Med Genet 1999; 86: 130–133.
Uuspää V : Combined bilateral external ear deformity and hypoplastic mandible. Scand J Plast Reconstr Surg 1978; 12: 165–167.
Poswillo D : The pathogenesis of the first and second branchial arch syndromes. Oral Surg Oral Med Oral Pathol 1973; 35: 302–328.
Vendramini S, Richieri-Costa A, Guion-Almeida ML : Oculoauriculovertebral spectrum with radial defects: a new syndrome or an extension of the oculoauriculovertebral spectrum? Report of fourteen Brazilian cases and review of the literature. Eur J Hum Genet 2007; 15: 411–421.
The Treacher Collins Collaborative Group: Positional cloning of a gene involved in the pathogenesis of Treacher Collins syndrome. Nat Genet 1996; 12: 130–136.
Splendore A, Fanganiello RD, Masotti C, Morganti LSC, Passos-Bueno MR : TCOF1 mutation database: novel mutation in the alternatively spliced Exon 6A and update in mutation nomenclature. Hum Mutat 2005; 25: 429–434.
Fisher S, Ludecke HJ, Wieczorek D, Bohringer S, Gillessen-Kaesbach G, Horsthemke B : Histone acetylation dependent allelic expression imbalance of BAPX1 in patients with the oculo-auriculo-vertebral spectrum. Hum Mol Genet 2006; 15: 581–587.
Agulnik SI, Papaioannou VE, Silver LM : Cloning, mapping, and expression analysis of TBX15, a new member of the T-box gene family. Genomics 1998; 51: 68–75.
Gong SG, Gong TW, Shum L : Identification of markers of the midface. J Dent Res 2005; 84: 69–72.
Mitsiadis TA, Romeas A, Lendahl U, Sharpe PT, Farges JC : Notch2 protein distribution in human teeth under normal and pathological conditions. Exp Cell Res 2003; 282: 101–109.
Larsson C, Lardelli M, White I, Lendahl U : The human NOTCH1, 2, and 3 genes are located at chromosome positions 9q34, 1p13–p11, and 19p13.2–p13.1 in regions of neoplasia-associated translocation. Genomics 1994; 24: 253–258.
Acknowledgements
We gratefully acknowledge the patients and their relatives. We would also like to thank Constância Gotto for secretarial assistance, Karina Lezirovitz and Fernanda Jehee for technical support. This work was supported by grants from Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP/CEPID) and Conselho Nacional de Pesquisa (CNPq).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on European Journal of Human Genetics website (http://www.nature.com/ejhg)
Rights and permissions
About this article
Cite this article
Masotti, C., Oliveira, K., Poerner, F. et al. Auriculo-condylar syndrome: mapping of a first locus and evidence for genetic heterogeneity. Eur J Hum Genet 16, 145–152 (2008). https://doi.org/10.1038/sj.ejhg.5201955
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201955
Keywords
This article is cited by
-
Prenatal diagnosis of auriculocondylar syndrome with a novel missense variant of GNAI3: a case report
BMC Pregnancy and Childbirth (2021)
-
Novel variants in GNAI3 associated with auriculocondylar syndrome strengthen a common dominant negative effect
European Journal of Human Genetics (2015)

{kind=link}
{kind=link}
{kind=link}