
Abstract
Skeletal dysplasias are difficult to diagnose for the nonexpert. In a previous study of patients with multiple epiphyseal dysplasia (MED), we identified cartilage oligomeric matrix protein (COMP) mutations in only 36% of cases and suspected that the low-mutation detection rate was partially due to misdiagnosis. We therefore instituted a clinical–radiographic review system, whereby all cases were evaluated by a panel of skeletal dysplasia experts (European Skeletal Dysplasia Network). Only those patients in whom the diagnosis of MED was confirmed by the panel were screened for mutations. Under this regimen the mutation detection rate increased to 81%. When clinical–radiological diagnostic criteria were relaxed the mutation rate dropped to 67%. We conclude that expert clinical–radiological review can significantly enhance mutation detection rates and should be part of any diagnostic mutation screening protocol for skeletal dysplasias.
Similar content being viewed by others


Introduction
Multiple epiphyseal dysplasia (MED) is a skeletal dysplasia characterized by delayed and irregular ossification of the epiphyses and early-onset osteoarthritis. Patients typically present with joint pain and stiffness particularly in the hips and knees and may have mild short stature.
MED can result from mutations in the genes encoding cartilage oligomeric matrix protein (COMP), type IX collagen (COL9A1, COL9A2 and COL9A3), matrilin-3 (MATN3) and the diastrophic dysplasia sulphate transporter (DTDST) (SLC26A2).1, 2 The frequency of mutations in these genes is not well established but previous studies have suggested detection rates of 10–28% for COMP,3, 4 14% for DTDST,4 10% for MATN3 and 5–10% for the type IX collagen genes.1, 4
In a recent study of 58 families with MED, we identified COMP mutations in 36% (21/58) of patients.5 These patients were diagnosed by their referring physician, mostly clinical geneticists. Even though some of the cases might have been caused by mutations in the other genes associated with MED, we suspected that misdiagnosis contributed to the low COMP mutation detection rate. We therefore tested the hypothesis that the preselection of cases, through expert clinical and radiological review, could significantly increase the mutation detection rate in MED.
Methods
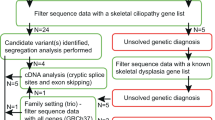
During an 18-month period between September 2003 and February 2005, 35 patients were referred to the European Skeletal Dysplasia Network (ESDN; www.esdn.org) with a suspected diagnosis of MED. Appropriate clinical descriptions and radiographs from each patient were reviewed by the ESDN Clinical and Radiographic Review Group. If the diagnosis of MED was confirmed by the expert panel, mutation screening of the known MED genes was offered. Patients who did not present the typical clinical–radiological findings of MED but were considered possible MED ‘variants’ were also offered mutation screening. Patients who did not present the clinical–radiological findings of MED and were not considered variants were either given an alternative diagnosis or classified as ‘unknown’.
Mutation screening of the COMP, COL9A1, COL9A2, COL9A3, MATN3 and DTDST genes was performed as described previously.5, 6, 7 Briefly, polymerase chain reaction (PCR) amplification of exons 8–19, including the immediate splice donor and acceptor sites of the COMP gene was performed using exon-specific intronic primers in eight separate PCR amplicons. PCR amplification of exon 2 of MATN3 was performed as a single reaction. Purified PCR products were used as a template for bi-directional fluorescent DNA sequencing and the DNA sequence data was compared against a reference sequence to detect variants using the Staden sequence analysis package.8 All DNA sequence traces were compared with a normal nonaffected control and also checked against the published cDNA sequences. One patient was diagnosed with a type II collagenopathy by the expert panel (see Results) and mutation screening of the COL2A1 was performed.
Results
Of the 35 patients referred to ESDN, 24 were diagnosed with ‘classical’ MED by the expert panel and five cases were considered possible MED variants (see Figures 1 and 2 for representative examples). Two patients were diagnosed with a type II collagenopathy and spondyloepimetaphyseal dysplasia (SEMD) with multiple dislocations (type Hall or leptodactylic type), respectively. No diagnosis could be made in four patients and these were classified as ‘unknown’.

Radiographs of a case diagnosed as ‘classical’ MED by the expert panel (ESDN-00080, 11 years old). The patient complained of joint pain and had mild-short stature. An Arg718Trp mutation in COMP was identified in this patient. Note small and irregular epiphyses in the knees, and flattening and irregular shape of the femoral heads.

Radiographs of a case diagnosed as a possible ‘MED variant’ by the expert panel (ESDN-00049, 16 years). The small size and irregular shape of the femoral head, and the flattened appearance of the femoral and tibial condyles are consistent with the post-pubertal changes seen in MED. The slender appearance of the long bones was not considered typical of MED. An Asp385Asn mutation in COMP was identified in this patient.
Genomic DNA for mutation analysis of the known ‘MED-disease’ genes could be obtained from 21 of the 29 eligible patients. Mutations were identified in 13 of the 16 patients with classical MED and in one of the five patients classified as possible MED variants. Of the 14 mutations identified, 13 were located in the COMP and one in the MATN3 gene. All mutations are listed in Table 1. In the patient diagnosed with a type II collagenopathy, a COL2A1 mutation was subsequently identified (patient AT-3844 in Hornaert et al9).
Three cases were considered to have typical features of MED but no mutation could be identified (Table 2). The radiographs and available clinical information of these cases are given in Figures 3, 4 and 5.

ESDN-00050: affected mother and son. Mother, 33 years, 1.68 m, two hip replacements. Radiographs show small and irregular femoral head (a), flattened appearance of the femoral and tibial condyles (b), slightly irregular vertebral bodies (c). Son 2.5 years, growing along the third centile. Radiograph shows slightly small femoral heads (d).

ESDN-00160: 50-year-old female who has suffered from painful knees and elbows since childhood. She has short stature (150 cm) and genu vara. Her father and six of her 12 siblings reportedly have short stature and varying degrees of hip and knee abnormalities. An affected sister had bilateral knee replacement at age 40. Radiographs show small but well-preserved femoral heads and flattened and irregularly shaped femoral condyles.

ESDN-00196: 4-year-old boy, normal height (98.5 cm). Radiographs show very small and irregular femoral heads and broad femoral necks, irregularly shaped epiphyses in the knee and a normal spine.
Discussion
By employing a clinical and radiographic review protocol, in which all referred MED cases were evaluated by a panel of skeletal dysplasia experts, we were able to increase the mutation detection rate to 81%. These findings indicate that mutations in the six known MED-disease genes are responsible for the vast majority of MED cases, and lower detection rates in previous studies are probably the result of misdiagnosis. Itoh et al10 recently reported a high mutation detection rate of 54% when screening patients with MED for mutations in the six known MED genes.10 These patients were recruited through specialized skeletal dysplasia clinics, which might have contributed to the high mutation detection rate. In our study, rigorous clinical–radiological preselection through a panel of experts resulted in an even higher mutation detection rate.
The lack of an identifiable mutation in 19% of cases with the typical clinical–radiological features of MED suggests that a small, but significant, proportion of MED cases are caused by mutation in an as yet unknown gene. It should be noted that our mutation screening strategy does not allow us to detect single exon deletions, more complex deletions or mutations in intronic splicing elements outside of the immediate splice donor and acceptor sequences. There remains therefore the remote possibility that mutations of this type could account for the phenotype in these cases.
Most mutations identified in this study were located in the COMP gene verifying that it is the major genetic locus for MED and suggesting that mutations in matrilin-3 and type IX collagen are responsible for only a small proportion of autosomal dominant MED cases. To date, all recessive forms of MED (rMED: EDM4) can be ascribed to mutations in SLC26A2, but this does not exclude the possibility that mutations in other genes can also account for rMED.
The relaxation of diagnostic criteria to allow for the existence of possible MED variants significantly decreased the mutation detection rate. In only one of five cases with an atypical presentation was a mutation identified (20%). These findings indicate that the MED phenotype is relatively well defined and that while the identification of rare MED variants is important from an academic point of view, strict diagnostic criteria should be applied in a routine diagnostic setting to allow for an efficient screening protocol.
In addition to improving the mutation detection rate and thus making genetic testing for MED more efficient, expert clinical–radiographic review allowed the revision of the diagnosis in two cases. One case was diagnosed with a type II collagenopathy, and a mutation in COL2A1 was subsequently identified. No molecular testing is available to confirm the revised diagnosis of SEMD (type Hall or leptodactylic type) in the second case, but the consensus of several experts makes this diagnosis very likely. Both cases would have remained undiagnosed without expert clinical–radiological review before genetic testing.
In summary, we conclude that expert clinical and radiological review can improve diagnostic accuracy and significantly enhance mutation detection rates. Review and preselection should be part of any diagnostic mutation screening protocol for patients with skeletal dysplasias or any other rare genetic disease that requires expert knowledge for diagnosis. Furthermore, our data confirm that COMP is the major genetic locus for autosomal dominant forms of MED.
References
Briggs MD, Chapman KL : Pseudoachondroplasia and multiple epiphyseal dysplasia: mutation review, molecular interactions, and genotype to phenotype correlations. Hum Mutat 2002; 19: 465–478.
Rossi A, Superti-Furga A : Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat 2001; 17: 159–171.
Unger SL, King LM, Sobetzko D, Superti-Furga A, Cohn DH : A multiplcity of loci for multiple epiphyseal dysplasia. Am J Hum Genet 2000; 67: 371.
Jakkula E, Makitie O, Czarny-Ratacjzak M et al: Mutations in the known genes are not the major cause of MED; distinctive phenotypic entities among patients with no identified mutations. Eur J Hum Genet 2005; 13: 292–301.
Kennedy J, Jackson G, Ramsden S et al: COMP mutation screening as an aid for the clinical diagnosis and counselling of patients with a suspected diagnosis of pseudoachondroplasia or multiple epiphyseal dysplasia. Eur J Hum Genet 2005; 13: 547–555.
Jackson GC, Barker FS, Jakkula E et al: Missense mutations in the beta strands of the single A-domain of matrilin-3 result in multiple epiphyseal dysplasia. J Med Genet 2004; 41: 52–59.
Superti-Furga A, Neumann L, Riebel T et al: Recessively inherited multiple epiphyseal dysplasia with normal stature, club foot, and double layered patella caused by a DTDST mutation. J Med Genet 1999; 36: 621–624.
Staden R, Beal KF, Bonfield JK : The Staden package, 1998. Methods Mol Biol 2000; 132: 115–130.
Hoornaert KP, Dewinter C, Vereecke I et al: The phenotypic spectrum in patients with arginine to cysteine mutations in the COL2A1 gene. J Med Genet 2006; 43: 406–413.
Itoh T, Shirahama S, Nakashima E et al: Comprehensive screening of multiple epiphyseal dysplasia mutations in Japanese population. Am J Med Genet A 2006; 140: 1280–1284.
Acknowledgements
The European Skeletal Dysplasia Network (ESDN: www.ESDN.org) is a combined Research and Demonstration project funded by the European Commission 5th Framework (QLG1-01-2001-02188). We thank our many clinical collaborators who referred cases to ESDN and for their continued support of the network. We acknowledge Drs B Kerr, S Smithson, SN Mohammed, K Ten Berg and PA Terhal, who provided the clinical description and radiographs shown in this paper.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Zankl, A., Jackson, G., Crettol, L. et al. Preselection of cases through expert clinical and radiological review significantly increases mutation detection rate in multiple epiphyseal dysplasia. Eur J Hum Genet 15, 150–154 (2007). https://doi.org/10.1038/sj.ejhg.5201744
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201744
Keywords
This article is cited by
-
Recessive multiple epiphyseal dysplasia (rMED) with homozygosity for C653S mutation in the DTDST gene - Phenotype, molecular diagnosis and surgical treatment of habitual dislocation of multilayered patella: Case report
BMC Musculoskeletal Disorders (2010)
-
Cartilage oligomeric matrix protein
AfCS-Nature Molecule Pages (2010)
