
Abstract
Recently, deletions encompassing the nuclear receptor binding SET-Domain 1 (NSD1) gene have been described as the major cause of Japanese patients with the Sotos syndrome, whereas point mutations have been identified in the majority of European Sotos syndrome patients. In order to investigate a possible phenotype–genotype correlation and to further define the predictive value of NSD1 mutations, we performed mutational analysis of the NSD1 gene in 20 patients and one familial case with Sotos syndrome, five patients with Weaver syndrome, six patients with unclassified overgrowth/mental retardation, and six patients with macrocephaly/mental retardation. We were able to identify mutations within the NSD1 gene in 18 patients and the familial case with Sotos syndrome (90%). The mutations (six nonsense, eight frame shifts, three splice site, one missense, one in-frame deletion) are expected to result in an impairment of NSD1 function. The best correlation between clinical assessment and molecular results was obtained for the Sotos facial gestalt in conjunction with overgrowth, macrocephaly, and developmental delay. In contrast to the high mutation detection rate in Sotos syndrome, none of the patients with Weaver syndrome, unclassified overgrowth/mental retardation and macrocephaly/mental retardation, harbored NSD1 mutations. We tested for large deletions by FISH analysis but were not able to identify any deletion cases. The results indicate that the great majority of patients with Sotos syndrome are caused by mutations in NSD1. Deletions covering the NSD1 locus were not found in the patients analyzed here.
Similar content being viewed by others

Introduction
Sotos syndrome is a well-defined and relatively common overgrowth syndrome characterized by pre- and postnatal overgrowth, developmental delay, advanced bone age, and a typical facial gestalt including macrodolichocephaly with frontal bossing, frontoparietal sparseness of hair, apparent hypertelorism, downslanting palpebral fissures, and facial flushing.
Weaver syndrome, another overgrowth syndrome, exhibits clinical signs that overlap with Sotos syndrome. However, the facial appearance including high and broad forehead, hypertelorism, prominent and long philtrum, and micrognathia is generally thought to be different from that of Sotos syndrome.1 The existence of rare cases with overlap between both syndromes and many phenotypic similarities have been interpreted as allelic rather than locus heterogeneity.2
Recently, the analysis of chromosomal rearrangements involving chromosome 5q35 in two patients with Sotos syndrome has identified a putative locus for the disease.3,4 Subsequently, deletions encompassing the nuclear receptor binding SET-Domain 1 (NSD1) gene as the major cause of Japanese patients with Sotos syndrome and point mutations as the major cause of European patients with Sotos syndrome have been published.5,6,7 Miyake et al8 found that microdeletions in Sotos syndrome mostly occurred in the paternally derived chromosome 5.
NSD1 belongs to a family of nuclear receptors (NR) that bind to DNA response elements upon binding of congate ligands such as steroid and thyroid hormones, or retinoids. NSD1 is a unique bifunctional cofactor with two distinct NR-interaction domains called NID−L and NID+L.9 NSD1 also contains several conserved functional domains, that is, SET, SAC, PWWP, and PHD. The SET domain (su(var)3-9, enhancer-of-zeste, trithorax) was first identified as a motif present in the Drosophila proteins SU(var)3-9, E(z) and TRx,10,11 and was subsequently found in a number of eucaryotic proteins. They were shown to play a role in cell growth and differentiation, to be associated with chromatin, and to function as a transcriptional represser and/or a transcriptional activator. NSD1 contains a Cys-rich region, which is composed of different arrangements of three conserved motifs, corresponding to a protein domain that has been called SAC for SET-associated Cys-rich domain.12 The SAC domain may have a function in chromosome binding. In addition to the SET and SAC domains, NSD1 contains six other domains including two proline–tryptophan–tryptophan–proline (PWWP) domains and five plant homeodomain protein (PHD) domains. It has been suggested that the PHD finger domains involve chromatin-mediated transcriptional regulation.12 The PWWP domain is thought to be involved in protein–protein interactions.13 Adjacent to the C-terminus of the PHD-V domain is another region rich in cysteines and histidines, possibly corresponding to a zinc-finger-like motif. The function of NSD1 remains largely unknown; however, the presence of activating as well as silencing domains and its property to bind liganded as well as unliganded NRs suggest that NSD1 could be a versatile NR intermediary factor controlling transcription either negatively or positively.12 The observation that haploinsufficiency of NSD1 induces overgrowth prompted Kurotaki et al5 to suggest that NSD1 acts as a corepressor of genes that promote growth.
In spite of the well-defined phenotype, Sotos syndrome is frequently difficult to diagnose particularly for the inexperienced clinician. Considerable overlap with other ill-defined overgrowth phenotypes exists, further complicating the situation. The molecular analysis allows an unequivocal classification and a better definition of the range of clinical manifestations in Sotos syndrome. The present study describes the results of mutation and microdeletion analysis of the NSD1 gene and the phenotypes in 20 patients and one familial case with Sotos syndrome, five patients with Weaver syndrome, six patients with an unclassified overgrowth/mental retardation syndrome, and in six patients with macrocephaly and mental retardation.
Patients
In all, 37 patients and one familial case with a tentative diagnosis of Sotos or Weaver syndrome or with other childhood overgrowth phenotypes were clinically assessed on the basis of anthropometric measurements, their photographs, bone age, and developmental delay. Phenotypic evaluation was undertaken separately by experienced syndromologists.
The clinical findings of the patients (ages ranging from birth to 37 years) are summarized in Table 1. In our series, three patients originated from one Turkish family (patients 16a, b, c) with an autosomal dominant pattern of inheritance and patient 16c died due to acute lymphoblastic leukemia. The others are sporadic cases of German and Turkish (patients 31, 32) origin. Prior to molecular analysis, the patients were phenotypically scored into four groups. Patients were considered having Sotos syndrome (n=21) if four of five major criteria, that is, overgrowth (height and weight >2 SD), macrocephaly (head circumference >2 SD), advanced bone age, developmental delay, and the Sotos facial gestalt, were present. Within this group, one patient (patient 3) showed craniofacial signs of both, Sotos and Weaver syndromes, and was therefore scored as an overlap phenotype. The diagnosis of Weaver syndrome (n=5) was established by the presence of the typical facial gestalt, characteristic growth pattern, accelerated bone age, and developmental delay. Patient 22 affected with Weaver syndrome had been reported previously (as patient 1).1 A group of six patients showed an unclassified phenotype with overgrowth, mental retardation, and a facial aspect different from that of Sotos or Weaver syndrome. The remaining six patients had macrocephaly and mental retardation.
Molecular analysis
Genomic DNA was extracted from blood lymphocytes using the commercial GenoPrepTM DNA Isolation Kit. All exons of NSD1 were amplified from genomic DNA with the primers designed by Kurotaki et al.5 (Matsumoto, personal communication). PCR reactions were performed in 20 μl containing 1 × PCR buffer, 1.5 or 2.5 mM MgC12, 100 μ M of each dNTP, 1 μl of each primer, and 2.5 U of Taq polymerase. The PCR conditions included an initial denaturation for 15 min at 96°C, followed by six cycles of 94°C for 30 s, 61–57°C for 45 s, 72°C for 60 s, 31 cycles of 94°C for 30 s, 55°C for 45 s, 72°C for 45 s, and a final extension for 10 min at 72°C. The analysis of PCR products was performed on 1.5% agarose gels.
PCR products were sequenced using Big Dye Terminator on an ABI Prism 3100 Genetic Analyzer (PE Biosystem, USA). Every mutation was confirmed by sequencing of the products from several independent PCRs.
Metaphase Fluorescence in situ hybridization
Metaphase chromosome preparations were obtained from PHA-stimulated lymphocyte cultures according to standard procedures. For fluorescence in situ hybridization (FISH) analysis, PAC clone RP1-118m12 was labeled with digoxigenin-11-dUTP by nick translation and preannealed with a 50-fold excess of Cot-1 DNA. Target slides were pretreated using standard methods. Hybridizations were carried out at 37°C overnight. Detection of the probes was achieved by using the fluorochrome-conjugated antibodies mouse-anti-digoxigenin–FITC, rabbit-anti-mouse–FITC, and goat-anti-rabbit–FITC.
Slides were counterstained with 4,6-diamidino-2-phenylindole. Images were obtained using an epifluorescence microscope (Axioscope; Zeiss Germany) equipped with a cooled CCD camera (Photometries) and analyzed using IP Lab Spectrum software.
Results
Identification of heterozygous mutations in NSD1
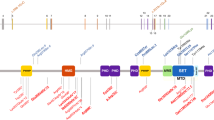
Among the patients examined, we were able to detect a total of 19 different heterozygous mutations (Figure 2 and Table 2) in 21 cases with Sotos syndrome. Of these mutations, 17 are novel and two have been reported previously.5,6 In the patient with the Sotos/Weaver overlap phenotype, we were able to identify a frame shift in exon 5. In the remaining 19 patients (two patients with Sotos syndrome, five patients with Weaver syndrome, six patients with unclassified overgrowth/mental retardation syndrome, and six patients with macrocephaly/mental retardation), no mutation was detected by direct sequencing.

Schematic representation of NSD1, showing the localization of mutations in patients with Sotos syndrome. The 23 exons of NSD1 are presented by boxes and introns are represented by lines. The domains of NSD1 are represented with different colored boxes. The mutations identified in the present study are indicated below the diagrammatic structure of NSD1.
Polymorphisms
The novel polymorphisms identified in this study were 352C>G (P118A), 1482C>T (C494C), 2242A>G (N748N), 3993T>C (D1331D), 4883T>C (M1628T), 6393C>G (V2131V), and 7636G>A (A2546T). The identified polymorphisms 1749G>A (E583E), 1792T>C (L599L), 1840T>G (V614L), 2071G>A (A691T), 2176T>C (S726P), 3106G>C (L1091I), 3705T>C (N1235N), 6750G>A (M2250I), 6782T>C (M2261T), 6829C>T (L2277L), and 6903G>C (G2301G) have been published previously.6
FISH investigations
A total of 19 patients (patients 1, 2, 3, 5, 6, 7, 8, 10, 11, 14, 15, 17, 18, 29, 30, 33, 34, 35, and 36) were investigated by FISH for a deletion of NSD1 locus using PAC RP1-118m12. The 5ptel probe (Appligene Oncor) was used as a control. In each of the 20 metaphases analyzed, FISH signals for the NSD1 locus were detected on both chromosomes 5. We tested PAC RP1-118m12 for its specificity by hybridizing digested PAC-DNA with probes specific for exons 2 and 4 of the NSD1 gene. FISH analysis could not be performed in the remaining patients because of the unavailability of blood samples.
Clinical data
We compared the clinical findings in the group of patients with Sotos syndrome and in the group of patients with Weaver syndrome, but not in the group of patients with unclassified overgrowth/mental retardation and with macrocephaly/mental retardation because of clinical heterogeneity.
Pre- and postnatal overgrowth, macrocephaly, the typical facial appearance, and development delay were all present in the majority of Sotos syndrome patients. However, in a subgroup of patients, either increased height or macrocephaly were lacking. Patients 10 and 16c were diagnosed as having Sotos syndrome on the basis of their facial gestalt, macrocephaly, and developmental delay, but their heights were still in the normal range at the age of 5 years (+2.0 SD) and of 5 years (+1.9 SD). In patients 9, 12, and 13, overgrowth, typical facial phenotype of Sotos syndrome, and development delay are present but macrocephaly was lacking (Table 1). Among our patients with Sotos syndrome, postnatal height varied from +1.9 to +6.3 SD. Head circumference ranged from +0.7 to +6.3 SD. In our series of patients with Sotos syndrome, intellectual impairment varied from very mild (patients 7, 16a) to severe (patients 5, 8).
Three patients with Sotos syndrome did not fulfill the criterion of accelerated bone age, patients 5 and 16c showed a bone age within the normal range at the age of 30 months and 5 years; in patient 6, aged 25 months, a retarded bone age has been determined.
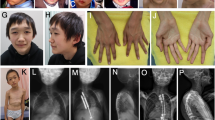
In patient 3, the diagnosis of Weaver syndrome was considered by the presence of the typical facial features during infancy, whereas in childhood the facial anomalies corresponded more to Sotos syndrome. Radiological assessment at 11 months of age showed no differences between the acceleration of carpal bones and phalangeal, radial, and ulnar epiphyses. In our opinion, the facial appearance of this patient draws attention to the existence of an overlapping phenotype between these syndromes (Figure 1h).

Facial photographs of patients harboring NSD1 mutations. (a–g) Patients with Sotos syndrome (a, patient 14; b, patient 5; c, patient 16b; d, patient 16a; e, patient 2; f, patient 13; g, patient 9). (h) Patient 3 with craniofacial symptoms of both Sotos and Weaver syndromes.
Besides overgrowth (ranged from +3.0 to +6.4 SD) and macrocephaly (ranged from +2.4 to +3.9 SD), the typical facial phenotype and developmental delay were present in all five patients with Weaver syndrome. Radiologically, patients 22, 24, 25, and 26 showed that the carpal bone age was more accelerated than the phalangeal bone age further supporting the diagnosis of Weaver syndrome.
The presence of NSD1 mutations was exclusively demonstrated in the group of patients with Sotos syndrome. In this group, the detection rate was 90%. The best correlation between clinical assessment and molecular results was obtained for the Sotos facial gestalt in conjunction with overgrowth, macrocephaly and developmental delay.
Discussion
We have conducted mutation and microdeletion analyses of the NSD1 gene in a series of 21 cases including one familial case with Sotos syndrome, five patients with Weaver syndrome, six patients with an unclassified overgrowth/mental retardation phenotype, and six patients with macrocephaly associated with mental retardation. All together, 19 mutations were detected that include frame shift, nonsense, missense, splice sites mutations, and in-frame deletion. In total, 17 mutations are described for the first time (Table 2, Figure 2). All were exclusively found in the group of patients with Sotos syndrome, indicating a very good correlation between the presence of NSD1 mutations and the Sotos phenotype. Mutations occurred throughout the gene, but the majority is clustered between exons 5 and 23. In our series, all mutations were identified only once. Two mutations (3536delA and R498X) were previously described in two patients with Sotos syndrome reported by Kurotaki et al5 and Douglas et al.6
The proportion of NSD1 aberrations among the group of patients affected with Sotos syndrome is 90% in comparison to the frequency (77, 76%) reported previously by Kurotaki et al5 and Douglas et al.6 Interestingly, the best correlation between the molecular and the clinical findings was achieved for the facial gestalt in conjunction with overgrowth, macrocephaly, and developmental delay. Mutations were identified in 19 of 21 cases with the typical Sotos syndrome face resulting in a predictive value for this clinical sign of about 90%. This result includes one patient (patient 3) with an overlapping facial phenotype of both, Sotos and Weaver syndromes.
Previous studies have described the overall frequency of 84% for advanced bone age in patients with Sotos syndrome.14 Our results confirm this finding and show that accelerated bone age is not an obligate symptom of Sotos syndrome.
In the patients with Weaver syndrome studied here, no NSD1 alterations were identified. Douglas et al6 identified three mutations in Weaver syndrome patients within a 40 amino-acid region encoding for the PHD-V domain and an adjacent domain rich in cysteines and histidines raising the possibility of a phenotype–genotype correlation. However, a mutation in this region was detected in a patient with typical the Sotos syndrome.6 The molecular results in Weaver syndrome patients reported here and in the overlapping case do not support this hypothesis. Furthermore, the familial case with Sotos syndrome (Figure 3) in our series has an in frame three amino-acid deletion removing one of the Cys residues between the PHD-V and the Cys/His-rich domain. This mutation is in close vicinity (four amino acids) to a published Weaver syndrome (patient COG62)6 mutation that replaces the cysteine residue 2183 by serine and is likely to have similar functional consequences. The function of the Cys/His-rich domain harboring the mutation is unknown, but sequence comparisons indicate homology to a zinc-finger-like motif. The three amino-acid deletion in our familial case removes one of the Cys residues and would be expected to have critical effects on the functional property of this domain. Alternatively, it may interfere with the function of the nearby PHD-V domain. The PHD-V domain contains a plant homeodomain, also designated as the C4HC3 motif,15 with a zinc-finger-like motif that predominantly occurs in proteins that function at the chromatin level.

Pedigree of the familial case.
In summary, no consistent relationship has been observed between specific mutations and the severity of the disease or the expression of a particular clinical sign. In two previous studies so far, 37 mutations of the NDS1 gene have been identified. Results from the present study, coupled with data from previously reported mutations 5,6,16 bring the total number of different mutations to 54. These data demonstrate that the NSD1 gene is subject of strong allelic heterogeneity, and there appears to be no major mutational hotspot in Sotos syndrome.
Familial cases are rare in Sotos syndrome raising the question of whether an underlying defect in fertility accounts for the paucity of familial cases. In the report of Douglas et al,6 the only patient who has a family history of Sotos syndrome showed a missense mutation in the PWWP-II domain raising the possibility that the observed mutation results in Sotos syndrome but not in reproductive impairment. Our molecular result of the familial case and the early truncating mutation in another familial case reported by Hoglund et al16 are not substantiated a correlation between the site and type of the mutation and fertility.
In contrast to the results of Kurotaki et al5 and Douglas et al6 deletions of the NSD1 gene were not found in the series of the 19 patients analyzed here. Using FISH analysis, Kurotaki et al5 reported a common 2.2-Mb deletion in 19/42 patients (66%) with Sotos syndrome, while in the studies described by Douglas et al6 a whole gene deletion occurred only in 3/37 patients (8%) with Sotos syndrome. Two of these patients showed deletion breakpoints within the above-mentioned interval detected by microsatellite analysis. The discrepancy between the Japanese and the European findings are difficult to explain and may be due to a selection bias. Using FISH analysis with a PAC clone, as described here, small deletions cannot be detected and may thus have been missed.
NSD1 aberrations were not identified in six patients with an unclassified overgrowth/mental retardation phenotype, nor six patients with macrocephaly associated with mental retardation. However, additional patients with nonspecific overgrowth syndromes need to be screened before the spectrum of phenotypes associated with NSD1 mutations becomes clear.
In our series, intragenic NSD1 mutations cause Sotos syndrome in the vast majority of patients clinically identified to have Sotos syndrome. NSD1 mutations may also be present in patients with Sotos syndrome lacking either macrocephaly or increased height. Aberrations of the NSD1 gene were not found associated with other overgrowth phenotypes such as Weaver syndrome. In contrast to prior reports, whole gene deletions could not be detected by FISH analysis.
References
Majewski F, Ranke M, Kemperdick H, Schmidt E : The Weaver syndrome: a rare type of primordial overgrowth. Eur J Pediatr 1981; 137: 277–282.
Opitz JM, Weaver DW, Reynolds Jr JF : The syndromes of Sotos and Weaver: reports and review. Am J Med Genet 1998; 79: 294–304.
Maroun C, Schmerler S, Hutcheon RG : Child with Sotos phenotype and a 5:15 translocation. Am J Med Genet 1994; 50: 291–293.
Imaizumi K, Kimura J, Matsuo M et al: Sotos syndrome associated with a de novo balanced reciprocal translocation t(5;8)(q35;q24.1). Am J Med Genet 2002; 107: 58–60.
Kurotaki N, Imaizumi K, Harada N et al: Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet 2002; 30: 365–366.
Douglas J, Hanks S, Temple IK et al: NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet 2003; 72: 132–143.
Nagai T, Matsumoto N, Kurotaki N et al: Sotos syndrome and haploinsufficiency of NSD1: clinical features of intragenic mutations and submicroscopic deletions. J Med Genet 2003; 40: 285–289.
Miyake N, Kurotaki N, Sugawara H et al: Preferential paternal origin of microdeletions caused by prezygotic chromosome or chromatid rearrangements in Sotos syndrome. Am J Hum Genet 2003; 72: 1331–1337.
Kurotaki N, Harada N, Yoshiura K, Sugano S, Niikawa N, Matsumoto N : Molecular characterization of NSD1, a human homologue of the mouse Nsd1 gene. Gene 2001; 279: 197–204.
Jones RS GW : The Drosophila polycomb-group gene Enhancer of zeste contains a region with sequence similarity to trithorax. Mol Cell Biol 1993; 13: 6357–6366.
Tschiersch B HA, Krauss V, Dorn R, Korge G, Reuter G : The protein encoded by the Drosophila position – effect variegation suppressor gene Su(var)3–9 combines domains of antagonistic regulators of homeotic gene complexes. EMBO J 1994; Aug 15; 13: 3822–3831.
Huang N, vom Baur E, Gamier JM et al: Two distinct nuclear receptor interaction domains in NSD1 a novel SET protein that exhibits characteristics of both corepressors and coactivators. EMBO J 1998; 17: 3398–3412.
Stec I, Nagl SB, van Ommen G–JB, den Dunnen JT : The PWWP domain: a potential protein–protein interaction domain in nuclear proteins influencing differentiation? FEBS Lett 2000; 473: 1–5.
Cole TR, Hughes HE : Sotos syndrome: a study of the diagnostic criteria and natural history. J Med Genet 1994; 31: 20–32.
Aasland R, Gibson TJ, Stewart AF : The PHD finger: implications for chromatin-mediated transcriptional regulation. Trends Biochem Sci 1995; 20: 56–59.
Hoglund P, Kurotaki N, Kytola S, Miyake N, Somer M, Matsumoto N : Familial Sotos syndrome is caused by a novel 1 bp deletion of the NSD1 gene. J Med Genet 2003; 40: 51–54.
Acknowledgements
We are grateful to all the patients and their families whose participation made this study possible.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Türkmen, S., Gillessen-Kaesbach, G., Meinecke, P. et al. Mutations in NSD1 are responsible for Sotos syndrome, but are not a frequent finding in other overgrowth phenotypes. Eur J Hum Genet 11, 858–865 (2003). https://doi.org/10.1038/sj.ejhg.5201050
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5201050
Keywords
This article is cited by
-
Structural and functional specificity of H3K36 methylation
Epigenetics & Chromatin (2022)
-
Drosophila NSD deletion induces developmental anomalies similar to those seen in Sotos syndrome 1 patients
Genes & Genomics (2021)
-
Chiari 1 malformation and exome sequencing in 51 trios: the emerging role of rare missense variants in chromatin-remodeling genes
Human Genetics (2021)
-
A novel mutation of NFIX causes Sotos-like syndrome (Malan syndrome) complicated with thoracic aortic aneurysm and dissection
Human Genome Variation (2017)
-
Clinical and genetic spectrum of 18 unrelated Korean patients with Sotos syndrome: frequent 5q35 microdeletion and identification of four novel NSD1 mutations
Journal of Human Genetics (2013)
