
Abstract
Leber's congenital amaurosis (LCA) is the most common cause of inherited childhood blindness and is characterised by severe retinal degeneration at or shortly after birth. We have identified a new locus, LCA9, on chromosome 1p36, at which the disease segregates in a single consanguineous Pakistani family. Following a whole genome linkage search, an autozygous region of 10 cM was identified between the markers D1S1612 and D1S228. Multipoint linkage analysis generated a lod score of 4.4, strongly supporting linkage to this region. The critical disease interval contains at least 5.7 Mb of DNA and around 50 distinct genes. One of these, retinoid binding protein 7 (RBP7), was screened for mutations in the family, but none was found.
Similar content being viewed by others

Introduction
Leber's congenital amaurosis (LCA) is the name given to a group of recessively inherited retinal dystrophies representing the most common genetic cause of blindness in infants and children. Symptoms include severe visual impairment at or within a few months of birth, nystagmus, sluggish pupils and an absent or reduced electroretinogram.1 Currently, six genes have been identified in which mutations are implicated in the pathogenesis of LCA,2,3,4,5,6,7 and two further loci are known to exist from linkage studies.8,9
The rationale for this study was that the level of genetic heterogeneity in LCA would be comparable to that observed with retinitis pigmentosa and thus there should be further unidentified loci. Linkage analysis in consanguineous pedigrees, known as homozygosity or autozygosity mapping, is a powerful approach to identify recessively inherited disease gene loci.10 Cultural precedents in some Pakistani communities have led to a high frequency of first-cousin marriages.11 Such families have a higher level of genetic disease than those in Western populations,12 making them an important resource in the quest to identify the genes that underlie recessively inherited human disease. We identified 11 members from two generations of a large consanguineous family affected by LCA (Figure 1), who originate from the Punjab region of Pakistan. Having excluded linkage to the known LCA loci, we then performed a whole genome linkage search to identify a new locus for LCA in this family.

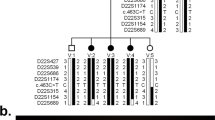
Pedigree of the LCA9 family. Microsatellite genotypes that span the autozygous region on chromosome 1p36 are shown in chromosomal order distal (top) to proximal (bottom). The boxed genotypes indicate the autozygous region in the affected individuals, and markers within the minimal disease region are shown in bold.
Materials and methods
All family members were subjected to a full ophthalmic examination by two independent ophthalmologists. The six affected individuals, whose ages ranged from 13 to 21 years, all gave a typical history of nonprogressive severe visual impairment since birth and photophobia. Best-corrected visual acuity was ‘perception of light’ in all affected eyes. Examination revealed nystagmus and posterior subcapsular lens opacities to be a universal finding. The youngest affected individual also had keratoconus. Examination of the posterior segment of the eye showed optic disc pallor, retinal vascular attenuation, varying degrees of macular staphyloma, widespread pigmentary disturbance heaviest equatorially and numerous fundal white spots. ERG testing and fundus photography were not possible as examinations were carried out in rural Pakistan. Nevertheless, all these features are consistent with a diagnosis of LCA.
Genomic DNA extraction was performed from peripheral blood leukocytes, using standard techniques. Genotyping was carried out using fluorescently labelled microsatellite markers assayed using an Applied Biosystems 377 instrument. Fragment length analysis was undertaken using ABI Prism Genescan and Genotyper 1.1.1 analysis packages.
Known LCA loci were excluded using the markers D1S3736 (RPE65), D1S1660 (CRB1), D6S968 (LCA5), D14S742 (RPGRIP), AGAT137 and GATA193A07 (LCA3), GATA158H04, D17S1308 and D17S974 (AIPL1 and GUCY2D), and D19S246 (CRX). Linkage analysis for the genome search was carried out using Genehunter v2.1,13 while the two- and three-point linkage analyses were produced using MLINK and LINKMAP, programmes from the Linkage suite.14 Mutation detection was carried out by direct sequencing of PCR products using the ABI BigDye™ v3.0 cycle sequencing kit. SSCP analysis was also carried out on the PCR products using the Pharmacia Biotech GeneGel Excel kit.
Results
Initially, genetic markers close to each of the known LCA loci were tested and linkage to these excluded in this family (data not shown). A whole genome linkage search was then carried out, whereby all 11 sampled members of the family were genotyped for the 407 markers in version 11 of the Marshfield screening set (http://www.marshfieldclinic.org/research/genetics/). The data generated from this were analysed for linkage using GeneHunter, assuming complete penetrance and a disease allele frequency of 1%. The allele frequencies used were those obtained in a panel of 20 Asian control individuals. To expedite the initial analysis, the consanguineous loops in the pedigree were not included. Exclusion was achieved with lod scores calculated to be −2 across most of the genome and no significant positive lod scores except in a region on chromosome 1p36, where a multipoint lod score of 3.5 was generated between the markers D1S1612 and D1S3669. To confirm linkage and define the extent of the locus, additional microsatellite markers were selected from the most recent genetic map available.15 By constructing haplotypes in this region, we were able to refine the locus to an autozygous region between the markers D1S1612 and D1S228 (Figure 1). Two-point lod scores for markers in this region are shown in Table 1. This region of 10 cM extends over approximately 5.7 Mb of DNA (UCSC genome browser June 2002 data freeze). Using a subset of these new data and a family structure incorporating the consanguineous loops with appropriate loop breaking, a three-point lod score of 4.4 was achieved with D1S2667 and D1S1597 using the programme LINKMAP (Figure 2).

Multipoint linkage analysis across the autozygous region on chromosome 1p36. Lod scores for each of the intervals were generated as a series of three-point calculations using LINKMAP. The marker order and distances CATC051-9cM-D1S1612-8.5cM-D1S2667-5cM-D1S1597-7cM-D1S3669-10cM-ATA47D07-16cM-ATA79C10 were taken primarily from the ‘Decode’ genetic map,15 but also used in conjunction with the latest Marshfield map.
Examination of the autozygous region using the UCSC genome browser revealed 46 candidate genes. One of these was retinoid binding protein 7 (RBP7). This gene consists of four exons extending over approximately 19 kb of genomic DNA. The four coding exons were identified using the UCSC genome browser and these were screened for mutations using both direct sequencing and SSCP with primers at least 50 bp into the surrounding intron sequences, but none were identified.
Discussion
At the time of writing, the critical region contained approximately 5.7 Mb of DNA, although the sequence assembly in this region contains several gaps. Until it is completed, the absolute order of markers and DNA content in this region will remain ill-defined. A total of 46 complete annotated genes lie in the critical region, one of which was screened in this study. The RBP7 gene codes for a protein that is a member of the retinoid binding protein family. The supply and processing of vitamin A derivatives is extremely important to normal retinal function16 and the related gene RBP4 has been found to be mutated in a recessively inherited degeneration of the retinal pigment epithelium,17 thus, RBP7 is a good candidate for the LCA9 gene. However, we failed to identify any mutations within its coding sequence. This does not completely exclude this gene, because mutations in the surrounding region that could potentially alter its expression would have been missed. Apart from the documented genes in the interval, there are also likely to be other unidentified genes. In light of the large number of candidate genes, progress with this project will depend initially on sampling of further members of the affected family and on the identification of other LCA families linked to this locus.
It is interesting to note that a mouse retinal degeneration locus (RD4) has been mapped to a region on mouse chromosome 4 which is orthologous to human chromosome 1p36.18,19 The phenotype associated with the RD4 mutation is dominantly inherited and is the result of a chromosomal inversion. The phenotype has an early onset like that of LCA, although the mutation is lethal when homozygous,19 whereas no other symptoms are observed in patients with LCA. Also of note is the fact that retinal degeneration is not a feature in patients with the 1p36 deletion syndrome, whereas anterior segment disorders are.20
In conclusion we have identified a new locus for Leber's congenital amaurosis located at chromosome 1p36 (7–14 Mb from telomere). This will facilitate genetic counselling for LCA families linked to this locus and will eventually lead to the identification of a gene that clearly has a critical role in the development and maintenance of the retina.
References
Foxman SG, Heckenlively JR, Bateman JB, Wirtschafter JD : Classification of congenital and early onset retinitis pigmentosa. Arch Ophthalmol 1985; 103: 1502–1506.
Freund CL, Wang QL, Chen S et al: De novo mutations in the CRX homeobox gene associated with Leber congenital amaurosis. Nat Genet 1998; 18: 311–312.
Perrault I, Rozet JM, Calvas P et al: Retinal-specific guanylate cyclase gene mutations in Leber's congenital amaurosis. Nat Genet 1996; 14: 461–464.
Marlhens F, Bareil C, Griffoin JM et al: Mutations in RPE65 cause Leber's congenital amaurosis. Nat Genet 1997; 17: 139–141.
Sohocki MM, Bowne SJ, Sullivan LS et al: Mutations in a new photoreceptor-pineal gene on 17p cause Leber congenital amaurosis. Nat Genet 2000; 24: 79–83.
Dryja TP, Adams SM, Grimsby JL et al: Null RPGRIP1 alleles in patients with Leber congenital amaurosis. Am J Hum Genet 2001; 68: 1295–1298.
Lotery AJ, Jacobson SG, Fishman GA et al: Mutations in the CRB1 gene cause Leber congenital amaurosis. Arch Ophthalmol 2001; 119: 415–420.
Dharmaraj S, Li Y, Robitaille JM et al: A novel locus for Leber congenital amaurosis maps to chromosome 6q. Am J Hum Genet 2000; 66: 319–326.
Stockton DW, Lewis RA, Abboud EB et al: A novel locus for Leber congenital amaurosis on chromosome 14q24. Hum Genet 1998; 103: 328–333.
Lander ES, Botstein D : Homozygosity mapping: a way to map human recessive traits with the DNA of inbred children. Science 1987; 236: 1567–1570.
Wahab A, Ahmad M : Biosocial perspective of consanguineous marriages in rural and urban Swat, Pakistan. J Biosoc Sci 1996; 28: 305–313.
Zlotogora J : What is the birth defect risk associated with consanguineous marriages? Am J Med Genet 2002; 109: 70–71.
Kruglyak L, Daly MJ, Reeve-Daly MP, Lander ES : Parametric and nonparametric linkage analysis: a unified multipoint approach. Am J Hum Genet 1996; 58: 1347–1363.
Lathrop GM, Lalouel JM, Julier C, Ott J : Strategies for multilocus linkage analysis in humans. Proc Natl Acad Sci USA 1984; 81: 3443–3446.
Kong A, Gudbjartsson DF, Sainz J et al: A high-resolution recombination map of the human genome. Nat Genet 2002; 31: 241–247.
Bok D : Processing and transport of retinoids by the retinal pigment epithelium. Eye 1990; 4: 326–332.
Seeliger MW, Biesalski HK, Wissinger B et al: Phenotype in retinol deficiency due to a hereditary defect in retinol binding protein synthesis. Invest Ophthalmol Vis Sci 1999; 40: 3–11.
Danciger M, Hendrickson J, Rao N et al: Positional cloning studies of the Rd4 mouse retinal degeneration. Invest Ophthalmol Vis Sci 2000; 41: S203 (abstract).
Roderick TH, Chang B, Hawes NL, Heckenlively JR : A new dominant retinal degeneration (Rd4) associated with a chromosomal inversion in the mouse. Genomics 1997; 42: 393–396.
Shapira SK, McCaskill C, Northrup H et al: Chromosome 1p36 deletions: the clinical phenotype and molecular characterization of a common newly delineated syndrome. Am J Hum Genet 1997; 61: 642–650.
Acknowledgements
This work was funded by the Wellcome Trust (Grant Nos. 061682, 035535 and 062444). We wish to thank the family studied here for their participation and assistance.
Author information
Authors and Affiliations
Corresponding author
Additional information
The first two authors contributed equally to this publication
Rights and permissions
About this article
Cite this article
Keen, T., Mohamed, M., McKibbin, M. et al. Identification of a locus (LCA9) for Leber's congenital amaurosis on chromosome 1p36. Eur J Hum Genet 11, 420–423 (2003). https://doi.org/10.1038/sj.ejhg.5200981
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200981
