
Abstract
Most carriers of autosomal recessive spinal muscular atrophy (SMA) have only one copy of SMN1 because of SMN1 gene deletions or gene conversions from SMN1 to SMN2, which has only one base difference in coding sequence from SMN1. Using SMN gene dosage analysis, we determined the copy numbers of SMN1 and SMN2 in the general population as well as in SMA patients and carriers. Increased SMN1 copy number is associated with decreased SMN2 copy number in the general population; that is, SMN2 copy number was decreased to one or zero copies in 11 of 13 individuals with three or four copies of SMN1, whereas only 71 of 164 individuals with two copies of SMN1 had one or zero copies of SMN2 (P<0.01). SMN2 copy number was increased to three or four in a subset of SMN1 deletion/conversion carriers, and in most SMA patients with a milder phenotype. In conclusion, our data provide evidence that gene conversion from SMN2 to SMN1 occurs, and that SMN1 converted from SMN2 is present in the general population.
Similar content being viewed by others

Introduction
Spinal muscular atrophy (SMA) is an autosomal recessive disorder caused by mutations in the SMN1 gene (MIM# 600354).1 SMN2 (MIM# 601627), an SMN1 homologue, typically differs from SMN1 by five nucleotides (in intron 6, exon 7, intron 7, and noncoding exon 8).2 Only one of these nucleotide changes (840C→T) is in the coding sequence, and it is translationally silent.2 Approximately 94% of clinically typical SMA patients lack both copies of SMN1 exon 7,3 and most carriers have only one copy of SMN1 exon 7, as determined by SMN gene dosage analysis.4,5 In addition to large deletions that include the entire SMN1 gene, loss of SMN1 exon 7 can occur by gene conversion from SMN1 to SMN2.6 SMA type III patients have, on average, more SMN2 copies than SMA type II or type I patients,7,8,9 and hence more copies of SMN2 derived by conversion from SMN1. The copy number of SMN2 correlates with longer survival and inversely with disease severity.8,9 Although gene conversion from SMN1 to SMN2 had been demonstrated, gene conversion from SMN2 to SMN1 has not. We show herein that increased SMN1 copy number is associated with decreased SMN2 copy number in the general population, which provides evidence that gene conversion from SMN2 to SMN1 occurs, and that SMN1 converted from SMN2 is present in the general population.
Materials and methods
Sample collection and DNA extraction
Using Puregene reagents (Gentra Systems, Minneapolis, MN, USA), DNA was extracted from peripheral blood specimens that were received with informed consent by the Molecular Pathology Laboratory of the Hospital of the University of Pennsylvania for SMN analyses on a clinical basis. We used results from all available individuals with no family history of SMA, (N=180)10 and 107 parents of an SMA patient; either parents with only one copy of SMN1, or parents of an SMA child who lacked SMN1 or had only one copy of SMN1 with a high clinical index of suspicion for SMN1-related SMA. Results were anonymized and used for our study. We also randomly selected and anonymized samples from 32 SMA patients with a known clinical type for our study.
SMN1 and SMN2 copy number assay (SMN gene dosage analysis)
A nonradioisotopic SMN gene dosage assay,7 a modification of the original method of McAndrew et al,4 was modified further, and validated for the determination of SMN1 and SMN2 copy numbers.11,12 SMN1 exon 7 copy number is used as a surrogate for SMN1 gene copy number. Briefly, SMN1 exon 7, SMN2 exon 7, a genomic two-copy reference (CFTR exon 4), and SMN and CFTR internal standards were coamplified in a single reaction. The PCR products were incubated with DraI, which digests only the SMN2 products, but not the SMN1 products. Copy number of SMN1 or SMN2 per cell (or more precisely, per diploid genome) was determined by quantification of the digested PCR products, followed by normalization utilizing the genomic standard, the internal standards, and five control samples with known SMN1 and SMN2 copy numbers. The quantitative accuracy of this method was validated as described.7,11,12 All test samples were analyzed in duplicate.
Results
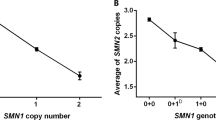
Distributions of SMN genotypes in unaffected individuals are shown in Table 1. SMN2 copy number and SMN1 copy number correlated inversely in the general population. SMN2 copy number was decreased to one or zero copies in 11 of 13 individuals with three or four copies of SMN1, whereas only 71 (43%) of 164 individuals with 2 SMN1 copies had one or zero SMN2 copies (P < 0.01; χ2 test).
There was a notable tendency for SMA type I carriers to have fewer SMN2 copies, compared to type II and type III carriers (Table 1). While only three (1.7%) of 177 noncarriers (in the column indicated by ‘No FH’ in Table 1) with two or more SMN1 copies had three SMN2 copies, SMN2 copy number was increased to three or four copies in 19 (17%) of 109 carriers with SMN1 deletion/conversion mutations (P < 0.001; χ2 test).
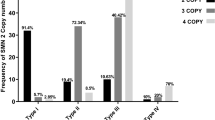
The distribution of SMN genotype in 32 SMA patients is as follows (with genotype expressed as ‘(SMN1 copy number):(SMN2 copy number)’): Among the 16 type patients, three were 0:1 and 13 were 0:2; the single type I-II patient was 0:2; among the eight type II patients, one was 0:2 and seven were 0:3 and one was 0.4; and one patient with very mild adult-onset SMA (which may be called type IV) was 0:4. The SMN2 copy number correlated inversely with disease severity.
Discussion
Using SMN gene dosage analysis, three or more copies of SMN1 were detected in some individuals in the general population, indicating the presence of chromosome 5s with two copies of SMN1.4,10 We meta-analyzed published data,3,4,8,10,13,14 and updated deduced SMN1 allele frequencies5 as follows: ‘zero-copy allele’ (chromosome 5 lacking SMN1 exon 7), 9.83 × 10−3; ‘one-copy allele’, 9.57 × 10−1; ‘two-copy allele’ (chromosome 5 with two copies of SMN1 exon 7), 3.27 × 10−2; and ‘1D allele’ (chromosome 5 with a small intragenic mutation in SMN1), 1.80 × 10−4.
One hypothesis to explain the presence of two copies of SMN1 on one chromosome 5 is unequal crossing over between homologous chromosomes during meiosis. Because of the presence of a large inverted repeat in the 5q13 region, and multiple smaller repeats contained therein, the SMN locus is considered highly susceptible to recombination. And, in fact, studies have shown that unequal crossing over at the SMN1 locus can cause de novo deletions of SMN1.7,15
An alternative hypothesis is that gene conversion from SMN2 to SMN1 can result in two SMN1 copies on one chromosome 5. In this scenario, SMN2 copy number would decrease after the gene conversion, in contrast to most unequal crossover events at the SMN1 locus. Our SMN1- and SMN2-copy-number data in the general population support this hypothesis. Our SMN2-copy-number data among SMA carriers and patients also support the hypo-thesis of the gene conversion from SMN1 to SMN2. Previous studies indicated that increased SMN2 copy number due to gene conversion from SMN1 to SMN2 is associated with a milder SMA phenotype.6,7,8,9
van der Steege et al16 studied three nucleotide differences in intron 6, exon 7, and exon 8, and found a hybrid SMN1/SMN2 gene (SMN1 intron 6/exon 7-SMN2 exon 8; intron 7 unknown) in one of their controls. Hahnen et al17 studied five nucleotide differences in intron 6, exon 7, intron 7, and exon 8, and found a hybrid gene (SMN1 intron 6/exon 7/intron 7-SMN2 exon 8) in one of their controls. The presence of these rare hybrid genes may support the hypothesis of gene conversions in either direction between SMN1 and SMN2, since a vast majority of hybrid genes present in SMA patients (presumably due to SMN1-to-SMN2 gene conversion6) had only SMN2 sequences except for one nucleotide in exon 8 (SMN2 intron 6/exon 7/intron 7-SMN1 exon 8).17 Other minor hybrid gene variants have also been described.17,18,19
In conclusion, our data provide population-based evidence of gene conversion from SMN2 to SMN1. Additional studies are necessary to confirm the hypothesis of gene conversion from SMN2 to SMN1.
References
Lefebvre S, Bürglen L, Reboullet S et al: Identification and characterization of a spinal muscular atrophy-determining gene. Cell 1995; 80: 155–165.
Bürglen L, Lefebvre S, Clermont O et al: Structure and organization of the human survival motor neurone (SMN) gene. Genomics 1996; 32: 479–482.
Wirth B : An update of the mutation spectrum of the survival motor neuron gene (SMN1) in autosomal recessive spinal muscular atrophy (SMA). Hum Mutat 2000; 15: 228–237.
McAndrew PE, Parsons DW, Simard LR et al: Identification of proximal spinal muscular atrophy carriers and patients by analysis of SMNT and SMNC gene copy number. Am J Hum Genet 1997; 60: 1411–1422.
Ogino S, Wilson RB : Genetic testing and risk assessment for spinal muscular atrophy (SMA). Hum Genet 2002; 111: 477–500.
Burghes AH : When is a deletion not a deletion? When it is converted [editorial]. Am J Hum Genet 1997; 61: 9–15.
Chen KL, Wang YL, Rennert H et al: Duplications and de novo deletions of the SMNt gene demonstrated by fluorescence-based carrier testing for spinal muscular atrophy. Am J Med Genet 1999; 85: 463–469.
Feldkötter M, Schwarzer V, Wirth R, Wienker TF, Wirth B : Quantitative analyses of SMN1 and SMN2 based on real-time LightCycler PCR: fast and highly reliable carrier testing and prediction of severity of spinal muscular atrophy. Am J Hum Genet 2002; 70: 358–368.
Taylor JE, Thomas NH, Lewis CM et al: Correlation of SMNt and SMNc gene copy number with age of onset and survival in spinal muscular atrophy. Eur J Hum Genet 1998; 6: 467–474.
Ogino S, Leonard DG, Rennert H, Ewens WJ, Wilson RB : Genetic risk assessment in carrier testing for spinal muscular atrophy. Am J Med Genet 2002; 110: 301–307.
Ogino S, Leonard DG, Rennert H, Gao S, Wilson RB : Heteroduplex formation in SMN gene dosage analysis. J Mol Diagn 2001; 3: 150–157.
Ogino S, Wilson RB : Quantification of PCR bias caused by a single nucleotide polymorphism in SMN gene dosage analysis. J Mol Diagn 2002; 4: 185–190.
Ogino S, Leonard DG, Rennert H, Wilson RB : Spinal muscular atrophy genetic testing experience at an academic medical center. J Mol Diagn 2002; 4: 53–58.
Wirth B, Herz M, Wetter A et al: Quantitative analysis of survival motor neuron copies: identification of subtle SMN1 mutations in patients with spinal muscular atrophy, genotype–phenotype correlation, and implications for genetic counseling. Am J Hum Genet 1999; 64: 1340–1356.
Wirth B, Schmidt T, Hahnen E et al: De novo rearrangements found in 2% of index patients with spinal muscular atrophy: mutational mechanisms, parental origin, mutation rate, and implications for genetic counseling. Am J Hum Genet 1997; 61: 1102–1111.
van der Steege G, Grootscholten PM, Cobben JM et al: Apparent gene conversions involving the SMN gene in the region of the spinal muscular atrophy locus on chromosome 5. Am J Hum Genet 1996; 59: 834–838.
Hahnen E, Schönling J, Rudnik-Schöneborn S, Zerres K, Wirth B : Hybrid survival motor neuron genes in patients with autosomal recessive spinal muscular atrophy: new insights into molecular mechanisms responsible for the disease. Am J Hum Genet 1996; 59: 1057–1065.
Velasco E, Valero C, Valero A, Moreno F, Hernandez-Chico C : Molecular analysis of the SMN and NAIP genes in Spanish spinal muscular atrophy (SMA) families and correlation between number of copies of cBCD541 and SMA phenotype. Hum Mol Genet 1996; 5: 257–263.
Cuscö I, Barcelo MJ, del Rio E et al: Characterisation of SMN hybrid genes in Spanish SMA patients: de novo, homozygous and compound heterozygous cases. Hum Genet 2001; 108: 222–229.
Acknowledgements
We thank the following individuals for supporting various aspects of our work: PE McAndrew, TW Prior, H Rennert, K-L Chen, H-J Dong, C Turino, and T Smith.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ogino, S., Gao, S., Leonard, D. et al. Inverse correlation between SMN1 and SMN2 copy numbers: evidence for gene conversion from SMN2 to SMN1. Eur J Hum Genet 11, 275–277 (2003). https://doi.org/10.1038/sj.ejhg.5200957
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ejhg.5200957
Keywords
This article is cited by
-
Detection of SMN1 to SMN2 gene conversion events and partial SMN1 gene deletions using array digital PCR
neurogenetics (2021)
-
Spinal muscular atrophy in Venezuela: quantitative analysis of SMN1 and SMN2 genes
Egyptian Journal of Medical Human Genetics (2020)
-
Transmission characteristics of SMN from 227 spinal muscular atrophy core families in China
Journal of Human Genetics (2020)
-
Validation of a high resolution NGS method for detecting spinal muscular atrophy carriers among phase 3 participants in the 1000 Genomes Project
BMC Medical Genetics (2015)
-
Universal fluorescent multiplex PCR and capillary electrophoresis for evaluation of gene conversion between SMN1 and SMN2 in spinal muscular atrophy
Analytical and Bioanalytical Chemistry (2010)
