
Abstract
Glycogen storage disease type II (GSD-II) is a lysosomal storage disorder in which the lack of human acid-alpha glucosidase (hGAA) activity results in massive accumulations of glycogen in cardiac and skeletal muscle fibers. Affected individuals die of cardiorespiratory failure secondary to the skeletal and/or cardiac muscle involvement. Recombinant hGAA enzyme replacement therapy (ERT) is currently in clinical trials and, although promising, ERT may be limited by large-scale production issues and/or the need for frequent infusions. These limitations could be circumvented or augmented by gene therapy strategies. Previous findings in our lab demonstrated that hepatic targeting of a modified adenovirus vector expressing human GAA was able to correct the glycogen accumulation in multiple affected muscles in the GAA-KO mice, by virtue of high-level, hepatic secretion of hGAA. However, although the vector persisted and expressed hGAA for 6 months in the liver, plasma hGAA was not detectable beyond 10 dpi (days postinjection), and reaccumulation of glycogen was observed. Two possibilities may have contributed to this phenomenon, the shut down of the CMV promoter and/or the onset of high levels of anti-hGAA antibodies. In order to test these and other possibilities, we have now developed an immune-deficient mouse model of GSD-II by interbreeding GAA-KO mice with severe combined immune-deficient (SCID) mice, generating double knockout, GAA-KO/SCID mice. In this new mouse model, we evaluated the efficacy of an [E1-, polymerase-] AdhGAA vector, in the absence of anti-hGAA antibody responses. After intravenous injection, GAA detection in the plasma was prolonged for at least 6 months secondary to the lack of anti-hGAA antibody production in all of the treated mice. GAA-KO/SCID mice treated with high doses of viral vector demonstrated longer durations of glycogen correction in both skeletal and cardiac muscles, relative to mice injected with lower doses of the vector. Notably, within 2 weeks of vector injection, muscle strength and coordination was normalized, and the improved muscle function persisted for at least 6 months. In summary, this new mouse model of GSD-II now makes it possible to assess the full potential for efficacy of any GAA-expressing vector (and/or ERT) contemplated for use in GSD-II gene therapy, without the negative influence that anti-hGAA antibodies entail.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

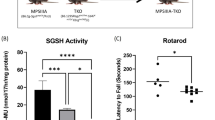
References
Raben N, Plotz P, Byrne BJ . Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr Mol Med 2002; 2: 145–166.
Wisselaar HA et al. Structural functional changes of lysosomal acid alpha-glucosidase during intracellular transport maturation. J Biol Chem 1993; 268: 2223–2231.
Hoefsloot LH, Hoogeveen-Westerveld M, Reuser AJ, Oostra BA . Characterization of the human lysosomal alpha-glucosidase gene. Biochem J 1990; 272: 493–497.
Reuser AJ et al. Glycogenosis type II (acid maltase deficiency). Muscle Nerve 1995; 3: S61–S69.
Chen YT, Amalfitano A . Towards a molecular therapy for glycogen storage disease type II (Pompe disease). Mol Med Today 2000; 6: 245–251.
Bijvoet AG et al. Human acid alpha-glucosidase from rabbit milk has therapeutic effect in mice with glycogen storage disease type II. Hum Mol Genet 1999; 8: 2145–2153.
Van den Hout H et al. Recombinant human alpha-glucosidase from rabbit milk in Pompe patients. Lancet 2000; 356: 397–398.
Van Hove JL et al. High-level production of recombinant human lysosomal acid alpha-glucosidase in Chinese hamster ovary cells which targets to heart muscle corrects glycogen accumulation in fibroblasts from patients with Pompe disease. Proc Natl Acad Sci USA 1996; 93: 65–70.
Amalfitano A et al. Systemic correction of the muscle disorder glycogen storage disease type II after hepatic targeting of a modified adenovirus vector encoding human acid-alpha-glucosidase. Proc Natl Acad Sci USA 1999; 96: 8861–8866.
Ding EY et al. Long-term efficacy after [E1-, polymerase-] adenovirus-mediated transfer of human acid-alpha-glucosidase gene into glycogen storage disease type II knockout mice. Hum Gene Ther 2001; 12: 955–965.
Ding E et al. Efficacy of gene therapy for a prototypical lysosomal storage disease (GSD-II) is critically dependent on vector dose, transgene promoter, and the tissues targeted for vector transduction. Mol Ther 2002; 5: 436–446.
Pauly DF et al. Intercellular transfer of the virally derived precursor form of acid alpha-glucosidase corrects the enzyme deficiency in inherited cardioskeletal myopathy Pompe disease. Hum Gene Ther 2001; 12: 527–538.
Raben N et al. Conditional tissue-specific expression of the acid alpha-glucosidase (GAA) gene in the GAA knockout mice: implications for therapy. Hum Mol Genet 2001; 10: 2039–2047.
McVie-Wylie AJ et al. Multiple muscles in the AMD quail can be cross-corrected of pathologic glycogen accumulation after intravenous injection of an [E1-, polymerase-] adenovirus vector encoding human acid-alpha-glucosidase. J Gene Med 2003; 5: 399–406.
Amalfitano A et al. Recombinant human acid alpha-glucosidase enzyme therapy for infantile glycogen storage disease type II: results of a phase I/II clinical trial. Genet Med 2001; 3: 132–138.
Raben N et al. Targeted disruption of the acid alpha-glucosidase gene in mice causes an illness with critical features of both infantile and adult human glycogen storage disease type II. J Biol Chem 1998; 273: 19086–19092.
Blunt T et al. Identification of a nonsense mutation in the carboxyl-terminal region of DNA-dependent protein kinase catalytic subunit in the SCID mouse. Proc Natl Acad Sci USA 1996; 93: 10285–10290.
Araki R et al. Nonsense mutation at Tyr-4046 in the DNA-dependent protein kinase catalytic subunit of severe combined immune deficiency mice. Proc Natl Acad Sci USA 1997; 94: 2438–2443.
Sandig V et al. Optimization of the helper-dependent adenovirus system for production and potency in vivo. Proc Natl Acad Sci USA 2000; 97: 1002–1007.
Kikuchi T et al. Clinical and metabolic correction of pompe disease by enzyme therapy in acid maltase-deficient quail. J Clin Invest 1998; 101: 827–833.
Fraites TJJ et al. Correction of the enzymatic and functional deficits in a model of Pompe disease using adeno-associated virus vectors. Mol Ther 2002; 5: 571–578.
Martin-Touaux E et al. Muscle as a putative producer of acid alpha-glucosidase for glycogenosis type II gene therapy. Hum Mol Genet 2002; 11: 1637–1645.
Sun B et al. Long-term correction of glycogen storage disease type II with a hybrid Ad-AAV vector. Mol Ther 2003; 7: 193–201.
Loser P, Jennings GS, Strauss M, Sandig V . Reactivation of the previously silenced cytomegalovirus major immediate-early promoter in the mouse liver: involvement of NfkappaB. J Virol 1998; 72: 180–190.
Raben N et al. Glycogen stored in skeletal but not in cardiac muscle in acid alpha-glucosidase mutant (Pompe) mice is highly resistant to transgene-encoded human enzyme. Mol Ther 2002; 6: 601–608.
Van der Ploeg AT et al. Receptor-mediated uptake of acid alpha-glucosidase corrects lysosomal glycogen storage in cultured skeletal muscle. Pediatr Res 1988; 24: 90–94.
Hesselink RP, Wagenmakers AJ, Drost MR, Van der Vusse GJ . Lysosomal dysfunction in muscle with special reference to glycogen storage disease type II. Biochim Biophys Acta 2003; 1637: 164–170.
Van der Ploeg AT et al. Intravenous administration of phosphorylated acid alpha-glucosidase leads to uptake of enzyme in heart skeletal muscle of mice. J Clin Invest 1991; 87: 513–518.
Martiniuk F et al. Extensive genetic heterogeneity in patients with acid alpha glucosidase deficiency as detected by abnormalities of DNA and mRNA. Am J Hum Genet 1990; 47: 73–78.
Acknowledgements
We thank AJ McVie-Wylie and S Lawson for technical support in the genotyping analysis of the GAA-KO allele. AA and YTC were both supported by the MDA (USA) and the Genzyme Corporation.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Xu, F., Ding, E., Liao, S. et al. Improved efficacy of gene therapy approaches for Pompe disease using a new, immune-deficient GSD-II mouse model. Gene Ther 11, 1590–1598 (2004). https://doi.org/10.1038/sj.gt.3302314
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.gt.3302314
Keywords
This article is cited by
-
Molecular Approaches for the Treatment of Pompe Disease
Molecular Neurobiology (2020)
-
Long-term neurologic and cardiac correction by intrathecal gene therapy in Pompe disease
Acta Neuropathologica Communications (2017)
-
Current and Future Treatments for Lysosomal Storage Disorders
Current Treatment Options in Neurology (2017)
-
Long-term, high-level hepatic secretion of acid α-glucosidase for Pompe disease achieved in non-human primates using helper-dependent adenovirus
Gene Therapy (2016)
