
Abstract
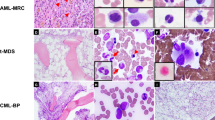
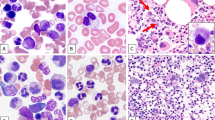
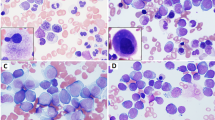
Myeloid sarcoma (MS) is a rare neoplasm whose knowledge is largely based on case reports and/or technically dated contributions. Ninety-two MSs in adulthood with clinical data available were evaluated both morphologically and immunohistochemically. Seventy-four cases were also studied by fluorescent in situ hybridization on tissue sections and/or conventional karyotyping on bone marrow or peripheral blood. Histologically, 50% of the tumors were of the blastic type, 43.5% either monoblastic or myelomonocytic and 6.5% corresponded to different histotypes. CD68/KP1 was the most commonly expressed marker (100%), followed by myeloperoxidase (83.6%), CD117 (80.4%), CD99 (54.3%), CD68/PG-M1 (51%), CD34 (43.4%), terminal-deoxy-nucleotidyl-transferase (31.5%), CD56 (13%), CD61/linker for activation of T cells (2.2%), CD30 (2.2%) and CD4 (1.1%). Foci of plasmacytoid monocyte differentiation were observed in intestinal cases carrying inv16. Chromosomal aberrations were detected in about 54% of cases: monosomy 7(10.8%), trisomy 8(10.4%) and mixed lineage leukemia-splitting (8.5%) were the commonest abnormalities, whereas t(8;21) was rare (2.2%). The behavior was dramatic irrespective of presentation, age, sex, phenotype and cytogenetics. Most if not all, long survivors received bone-marrow transplantation. The present report expands the spectrum of our knowledge showing that MS has frequent monoblastic/myelomonocytic differentiation, displays distinctive phenotypic profile, carries chromosomal aberrations other than t(8;21), and requires supra-maximal therapy.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
References
Jaffe ES, Harris NL, Stein H, Vardiman JW (eds). Pathology and Genetics. Tumours of Haematopoietic and Lymphoid Tissues. Lyon: IARC Press, 2001, pp 104–105.
Neiman RS, Barcos M, Berard C, Bonner H, Mann R, Rydell RE et al. Granulocytic sarcoma: a clinicopathologic study of 61 biopsied cases. Cancer 1981; 48: 1426–1437.
Menasce LP, Banerjee SS, Beckett E, Harris M . Extra-medullary myeloid tumour (granulocytic sarcoma) is often misdiagnosed: a study of 26 cases. Histopathology 1999; 34: 391–398.
Tsimberidou AM, Kantarjian HM, Estey E, Cortes JE, Verstovsek S, Faderl S et al. Outcome in patients with nonleukemic granulocytic sarcoma treated with chemotherapy with or without radiotherapy. Leukemia 2003; 17: 1100–1103.
Breccia M, Mandelli F, Petti MC, D'Andrea M, Pescarmona E, Pileri SA et al. Clinico-pathological characteristics of myeloid sarcoma at diagnosis and during follow-up: report of 12 cases from a single institution. Leuk Res 2004; 28: 1165–1169.
Ferry JA, Srigley JR, Young RH . Granulocytic sarcoma of the testis: a report of two cases of a neoplasm prone to misinterpretation. Mod Pathol 1997; 10: 320–325.
Oliva E, Ferry JA, Young RH, Prat J, Srigley JR, Scully RE . Granulocytic sarcoma of the female genital tract: a clinicopathologic study of 11 cases. Am J Surg Pathol 1997; 21: 1156–1165.
Corpechot C, Lemann M, Brocheriou I, Mariette X, Bonnet J, Daniel MT et al. Granulocytic sarcoma of the jejunum: a rare cause of small bowel obstruction. Am J Gastroenterol 1998; 93: 2586–2588.
McCluggage WG, Boyd HK, Jones FG, Mayne EE, Bharucha H . Mediastinal granulocytic sarcoma: a report of two cases. Arch Pathol Lab Med 1998; 122: 545–547.
Gorczyca W, Weisberger J, Seiter K . Colonic adenomas with extramedullary myeloid tumor (granulocytic sarcoma). Leuk Lymphoma 1999; 34: 621–624.
Au WY, Shek TW, Ma SK, Leung G, Ooi GC, Liang R et al. Myeloblastoma (chloroma) in leukemia: case 2. Meningeal granulocytic sarcoma (chloroma) in essential thrombocythemia. J Clin Oncol 2000; 18: 3996–3997.
Breccia M, Petti MC, Fraternali-Orcioni G, Monarca B, Latagliata R, D'Elia GM et al. Granulocytic sarcoma with breast and skin presentation: a report of a case successfully treated by local radiation and systemic chemotherapy. Acta Haematol 2000; 104: 34–37.
Ascani S, Piccaluga PP, Pileri SA . Granulocytic sarcoma of main biliary ducts. Br J Haematol 2003; 121: 534.
Breccia M, D'Andrea M, Mengarelli A, Morano SG, D'Elia GM, Alimena G . Granulocytic sarcoma of the pancreas successfully treated with intensive chemotherapy and stem cell transplantation. Eur J Haematol 2003; 70: 190–192.
Imamura T, Matsuo S, Yoshihara T, Chiyonobu T, Mori K, Ishida H et al. Granulocytic sarcoma presenting with severe adenopathy (cervical lymph nodes, tonsils, and adenoids) in a child with juvenile myelomonocytic leukemia and successful treatment with allogeneic bone marrow transplantation. Int J Hematol 2004; 80: 186–189.
Elenitoba-Johnson K, Hodges GF, King TC, Wu CD, Medeiros LJ . Extramedullary myeloid cell tumors arising in the setting of chronic myelomonocytic leukemia. A report of two cases. Arch Pathol Lab Med 1996; 120: 62–67.
Hancock JC, Prchal JT, Bennett JM, Listinsky CM . Trilineage extramedullary myeloid cell tumor in myelodysplastic syndrome. Arch Pathol Lab Med 1997; 121: 520–523.
Kasahara S, Tsurumi H, Hara T, Goto H, Moriwaki H . Idiopathic myelofibrosis developing isolated granulocytic sarcoma with der (1;7)(q10; p10) after splenectomy and finally transforming to acute myelogenous leukemia. Leuk Lymphoma 2000; 39: 427–433.
Cankaya H, Ugras S, Dilek I . Head and neck granulocytic sarcoma with acute myeloid leukemia: three rare cases. Ear Nose Throat J 2001; 80: 224–226, 228–229.
Suzer T, Colakoglu N, Cirak B, Keskin A, Coskun E, Tahta K . Intracerebellar granulocytic sarcoma complicating acute myelogenous leukemia: a case report and review of the literature. J Clin Neurosci 2004; 11: 914–917.
Szomor A, Baranyai F, Tornoczky T, Losonczy H . Penile chloroma in a patient with secondary acute myeloid leukemia. Eur J Haematol 2002; 68: 322.
Maeng H, Cheong JW, Lee ST, Yang WI, Hahn JS, Ko YW et al. Isolated extramedullary relapse of acute myelogenous leukemia as a uterine granulocytic sarcoma in an allogeneic hematopoietic stem cell transplantation recipient. Yonsei Med J 2004; 45: 330–333.
Quintanilla-Martinez L, Zukerberg LR, Ferry JA, Harris NL . Extramedullary tumors of lymphoid or myeloid blasts. The role of immunohistology in diagnosis and classification. Am J Clin Pathol 1995; 104: 431–443.
Roth MJ, Medeiros LJ, Elenitoba-Johnson K, Kuchnio M, Jaffe ES, Stetler-Stevenson M . Extramedullary myeloid cell tumors. An immunohistochemical study of 29 cases using routinely fixed and processed paraffin-embedded tissue sections. Arch Pathol Lab Med 1995; 119: 790–798.
Chang CC, Eshoa C, Kampalath B, Shidham VB, Perkins S . Immunophenotypic profile of myeloid cells in granulocytic sarcoma by immunohistochemistry. Correlation with blast differentiation in bone marrow. Am J Clin Pathol 2000; 114: 807–811.
Chen J, Yanuck III RR, Abbondanzo SL, Chu WS, Aguilera NS . c-Kit (CD117) reactivity in extramedullary myeloid tumor/granulocytic sarcoma. Arch Pathol Lab Med 2001; 125: 1448–1452.
Miettinen M, Lasota J . KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl Immunohistochem Mol Morphol 2005; 13: 205–220.
Traweek ST, Arber DA, Rappaport H, Brynes RK . Extramedullary myeloid cell tumors. An immunohistochemical and morphologic study of 28 cases. Am J Surg Pathol 1993; 17: 1011–1019.
Kurata H, Okukubo M, Fukuda E, Ichihashi M, Ueda M . Myeloid markers should be undertaken in cases of CD56 positivity to exclude granulocytic sarcoma. Br J Dermatol 2002; 147: 609–611.
Petrella T, Bagot M, Willemze R, Beylot-Barry M, Vergier B, Delaunay M et al. Blastic NK-cell lymphomas (agranular CD4+CD56+ hematodermic neoplasms): a review. Am J Clin Pathol 2005; 123: 662–675.
Schwyzer R, Sherman GG, Cohn RJ, Poole JE, Willem P . Granulocytic sarcoma in children with acute myeloblastic leukemia and t(8;21). Med Pediatr Oncol 1998; 31: 144–149.
Bonig H, Gobel U, Nurnberger W . Bilateral exopthalmus due to retro-orbital chloromas in a boy with t(8;21)- positive acute myeloblastic acute leukemia. Pediatr Hematol Oncol 2002; 19: 597–600.
Rubnitz JE, Raimondi SC, Halbert AR, Tong X, Srivastava DK, Razzouk BI et al. Characteristics and outcome of t(8;21)-positive childhood acute myeloid leukemia: a single institution's experience. Leukemia 2002; 16: 2072–2077.
Fiegl M, Rieger C, Braess J, Haferlach T, Schnittger S, Schoch C et al. Isolated epidural chloroma with translocation t(15; 17) successfully treated with chemotherapy and all-trans-retinoic acid. Br J Haematol 2003; 122: 688–689.
Cornfield DB, Sun G, Ahmed B . Granulocytic sarcoma associated with a der(7;12)(q10;q10). Cancer Genet Cytogenet 2005; 156: 89–91.
Deeb G, Baer MR, Gaile DP, Sait SN, Barcos M, Wetzler M et al. Genomic profiling of myeloid sarcoma by array comparative genomic hybridization. Genes Chromosomes Cancer 2005; 44: 373–383.
Nicci C, Ottaviani E, Luatti S, Grafone T, Tonelli M, Motta MR et al. Molecular and cytogenetic characterization of a new case of t(5;17)(q35;q21) variant acute promyelocytic leukemia. Leukemia 2005; 19: 470–472.
Park KU, Lee DS, Lee HS, Kim CJ, Cho HI . Granulocytic sarcoma in MLL-positive infant acute myelogenous leukemia: fluorescence in situ hybridization study of childhood acute myelogenous leukemia for detecting MLL rearrangement. Am J Pathol 2001; 159: 2011–2016.
Douet-Guilbert N, Morel F, Le Bris MJ, Sassolas B, Giroux JD, De Braekeleer M . Rearrangement of MLL in a patient with congenital acute monoblastic leukemia and granulocytic sarcoma associated with a t(1;11)(p36;q23) translocation. Leuk Lymphoma 2005; 46: 143–146.
Pulsoni A, Falcucci P, Anghel G, Ribersani M, Petrucci MT, Pescarmona E et al. Isolated granulocytic sarcoma of the skin in an elderly patient: good response to treatment with local radiotherapy and low-dose methotrexate. J Eur Acad Dermatol Venereol 2000; 14: 216–218.
Finnegan D, Jones F, McMullin M . Acute myeloid leukemia with concurrent myeloid sarcoma treated with autologous bone marrow transplantation: two illustrative cases and a literature review. Hematol Oncol 2005; 23: 133–135.
Piccaluga PP, Martinelli G, Rondoni M, Malagola M, Gaitani S, Isidori A et al. Gemtuzumab ozogamicin for relapsed and refractory acute myeloid leukemia and myeloid sarcomas. Leuk Lymphoma 2004; 45: 1791–1795.
Carella AM, Carlier P, Pungolino E, Resegotti L, Liso V, Stasi R et al. Idarubicin in combination with intermediate-dose cytarabine and VP-16 in the treatment of refractory or rapidly relapsed patients with acute myeloid leukemia. The GIMEMA Cooperative Group. Leukemia 1993; 7: 196–199.
Avvisati G, Lo Coco F, Diverio D, Falda M, Ferrara F, Lazzarino M et al. AIDA (all-trans retinoic acid + idarubicin) in newly diagnosed acute promyelocytic leukemia: a Gruppo Italiano Malattie Ematologiche Maligne dell'Adulto (GIMEMA) pilot study. Blood 1996; 88: 1390–1398.
Hoffman R BEJ, Shattil S, Furie B, Cohen HJ, Silberstein LE, McGlave P . Hematology Basic Principles and Practice, 4th edn, Elsevier Churchill Livingstone: London, 2005, pp 1099–1120.
Pileri SA, Roncador G, Ceccarelli C, Piccioli M, Briskomatis A, Sabattini E et al. Antigen retrieval techniques in immunohistochemistry: comparison of different methods. J Pathol 1997; 183: 116–123.
Haralambieva E, Kleiverda K, Mason DY, Schuuring E, Kluin PM . Detection of three common translocation breakpoints in non-Hodgkin's lymphomas by fluorescence in situ hybridization on routine paraffin-embedded tissue sections. J Pathol 2002; 198: 163–170.
Cook JR . Paraffin section interphase fluorescence in situ hybridization in the diagnosis and classification of non-hodgkin lymphomas. Diagn Mol Pathol 2004; 13: 197–206.
Weir BS, Hill WG . Estimating F-statistics. Annu Rev Genet 2002; 36: 721–750.
Muir WM . Estimation of response to selection and utilization of control populations for additional information and accuracy. Biometrics 1986; 42: 381–391.
Kaplan EL, Meier P . Non-parametric estimation from incomplete observation. JAMA 1958; 58: 457–461.
Mantel N . Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep 1966; 50: 163–170.
Cox DR . Regression models and life-tables. J Stat Soc 1982; 34: 187–220.
Zhang PJ, Barcos M, Stewart CC, Block AW, Sait S, Brooks JJ . Immunoreactivity of MIC2 (CD99) in acute myelogenous leukemia and related diseases. Mod Pathol 2000; 13: 452–458.
Chen VM, McIlroy K, Loui JP, Fay K, Ward C . Extramedullary presentation of acute leukaemia: a case of myeloid/natural killer cell precursor leukaemia. Pathology 2003; 35: 325–329.
Fickers M, Theunissen P . Granulocytic sarcoma with expression of CD30. J Clin Pathol 1996; 49: 762–763.
Vermi W, Facchetti F, Rosati S, Vergoni F, Rossi E, Festa S et al. Nodal and extranodal tumor-forming accumulation of plasmacytoid monocytes/interferon-producing cells associated with myeloid disorders. Am J Surg Pathol 2004; 28: 585–595.
Facchetti F, Vermi W, Santoro A, Vergoni F, Chilosi M, Doglioni C . Neoplasms derived from plasmacytoid monocytes/interferon-producing cells: variability of CD56 and granzyme B expression. Am J Surg Pathol 2003; 27: 1489–1492; author reply 1492–1493.
Byrd JC, Weiss RB . Recurrent granulocytic sarcoma. An unusual variation of acute myelogenous leukemia associated with 8;21 chromosomal translocation and blast expression of the neural cell adhesion molecule. Cancer 1994; 73: 2107–2112.
Psiachou-Leonard E, Paterakis G, Stefanaki K, Mikraki-Christou V, Haidas S . Cerebellar granulocytic sarcoma in an infant with CD56+ acute monoblastic leukemia. Leuk Res 2001; 25: 1019–1021.
Jarvinen M, Andersson LC, Virtanen I . K562 erythroleukemia cells express cytokeratins 8, 18, and 19 and epithelial membrane antigen that disappear after induced differentiation. J Cell Physiol 1990; 143: 310–320.
Turner JJ, Milliken S . A case of keratin-positive acute myeloid leukemia: a possible role for cytokeratin 19 as a specific epithelial marker. Pathology 2000; 32: 98–101.
Mitelman F, Heim S . Quantitative acute leukemia cytogenetics. Genes Chromosomes Cancer 1992; 5: 57–66.
Falini B, Mecucci C, Tiacci E, Alcalay M, Rosati R, Pasqualucci L et al. Cytoplasmic nucleophosmin in acute myelogenous leukemia with a normal karyotype. N Engl J Med 2005; 352: 254–266.
Casas S, Aventin A, Fuentes F, Vallespi T, Granada I, Carrio A et al. Genetic diagnosis by comparative genomic hybridization in adult de novo acute myelocytic leukemia. Cancer Genet Cytogenet 2004; 153: 16–25.
Dusenbery KE, Howells WB, Arthur DC, Alonzo T, Lee JW, Kobrinsky N et al. Extramedullary leukemia in children with newly diagnosed acute myeloid leukemia: a report from the Children's Cancer Group. J Pediatr Hematol Oncol 2003; 25: 760–768.
Xavier SG, Fagundes EM, Hassan R, Bacchi C, Conchon M, Tabak DG et al. Granulocytic sarcoma of the small intestine with CBFbeta/MYH11 fusion gene: report of an aleukaemic case and review of the literature. Leuk Res 2003; 27: 1063–1066.
Russell SJ, Giles FJ, Thompson DS, Scanlon DJ, Walker H, Richards JD . Granulocytic sarcoma of the small intestine preceding acute myelomonocytic leukemia with abnormal eosinophils and inv(16). Cancer Genet Cytogenet 1988; 35: 231–235.
Acknowledgements
This work was supported by Italian Association for Cancer Research (AIRC, Milan), Italian Ministry of University and Research (MIUR, Rome), Fondazione Cassa di Risparmio in Bologna, Fondazione della Banca del Monte e Ravenna (Bologna) and BolognAIL (Bologna).
Author information
Authors and Affiliations
Corresponding author
Additional information
Supplementary Information accompanies the paper on the Leukemia website (http://www.nature.com/leu)
Rights and permissions
About this article
Cite this article
Pileri, S., Ascani, S., Cox, M. et al. Myeloid sarcoma: clinico-pathologic, phenotypic and cytogenetic analysis of 92 adult patients. Leukemia 21, 340–350 (2007). https://doi.org/10.1038/sj.leu.2404491
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.leu.2404491
Keywords
This article is cited by
-
Myeloid sarcoma and pathological fracture: a case report and review of literature
International Journal of Hematology (2023)
-
Top Ten Lymphoproliferative Lesions Not to Miss When Evaluating Oral Ulcer Biopsies
Head and Neck Pathology (2023)
-
Myeloid sarcoma: more and less than a distinct entity
Annals of Hematology (2023)
-
Gene Mutations and Targeted Therapies of Myeloid Sarcoma
Current Treatment Options in Oncology (2023)
-
Clinical characteristics, treatment, and prognosis of 118 cases of myeloid sarcoma
Scientific Reports (2022)
