
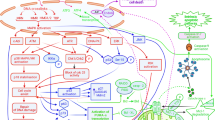
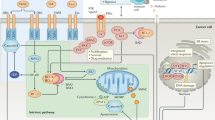
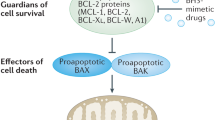
Abstract
Most chemotherapeutic drugs can induce tumor cell death by apoptosis. Analysis of the molecular mechanisms that regulate apoptosis has indicated that anticancer agents simultaneously activate several pathways that either positively or negatively regulate the death process. The main pathway from specific damage induced by the drug to apoptosis involves activation of caspases in the cytosol by pro-apoptotic molecules such as cytochrome c released from the mitochondrial intermembrane space. At least in some cell types, anticancer drugs also upregulate the expression of death receptors and sensitize tumor cells to their cognate ligands. The Fas-mediated pathway could contribute to the early steps of drug-induced apoptosis while sensitization to the cytokine TRAIL could be used to amplify the response to cytotoxic drugs. The Bcl-2 family of proteins, that includes anti- and pro-apoptotic molecules, regulates cell sensitivity mainly at the mitochondrial level. Anticancer drugs modulate their expression (eg through p53-dependent gene transcription), their activity (eg by phosphorylating Bcl-2) and their subcellular localization (eg by inducing the translocation of specific BH3-only pro-apoptotic proteins). Very early after interacting with tumor cells, anticancer drugs also activate lipid-dependent signaling pathways that either increase or decrease cell ability to die by apoptosis. In addition, cytotoxic agents can activate protective pathways that involve activation of NFκB transcription factor, accumulation of heat shock proteins such as Hsp27 and activation of proteins involved in cell cycle regulation. This review discusses how modulation of the balance between noxious and protective signals that regulate drug-induced apoptosis could be used to improve the efficacy of current therapeutic regimens in hematological malignancies.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others
References
Ross DD . Novel mechanisms of drug resistance in leukemia Leukemia 2000 14: 467–473
Wattel E, Solary E, Hecquet B, Caillot D, Ifrah N, Brion A, Mahé B, Milpied N, Janvier M, Guerci A, Rochant H, Cordonnier C, Dreyfus F, Buzyn A, Hoang-Ngoc L, Stoppa AM, Gratecos N, Sadoun A, Stamatoulas A, Tilly H, Brice P, Maloisel F, Lioure B, Desablens B, Pignon B, Abgrall JF, Leporrier M, Dupriez B, Guyotat D, Lepelley P, Fenaux P . Quinine improves the results of intensive chemotherapy in myelodysplatic syndromes expressing P-glycoprotein: results of a randomized study Br J Haematol 1998 102: 1015–1024
Solary E, Witz F, Caillot D, Moreau P, Desablens B, Cahn JY, Sadoun A, Berthou C, Maloisel F, Guyotat D, Casassus P, Ifrah N, Lamy B, Audhuy B, Colombat P, Harousseau JL . Combination of quinine as a potential reversing agent with mitoxantrone and cytarabine for the treatment of acute leukemias: a randomised multicentric study Blood 1996 88: 1198–1205
Martins LM, Mesner PW, Kottke TJ, Basi GS, Sinha S, Tung JS, Svingen PA, Madden BJ, Takahashi A, McCormick DJ, Earnshaw WC, Kaufmann SH . Comparison of caspase activation and subcellular localization in HL-60 and K562 cells undergoing etoposide-induced apoptosis Blood 1997 90: 4283–4296
Kroemer G, Reed JC . Mitochondrial control of cell death Nature Med 2000 6: 513–519
Zamzami N, Marchetti P, Castedo M, Zanin C, Vayssiere JL, Petit PX, Kroemer G . Reduction in mitochondrial potential constitutes an early irreversible step of programmed lymphocyte death in vivo J Exp Med 1995 181: 1661–1672
Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC . Inhibition of Bax channel-forming activity by Bcl-2 Science 1997 277: 370–372
Crompton M . The mitochondrial permeability transition pore and its role in cell death Biochem J 1999 341: 233–249
Schlesinger PH, Gross A, Yin XM, Yamamoto K, Saito M, Waksman G, Korsmeyer SJ . Comparison of the ion channel characteristics of proapoptotic BAX and antiapoptotic BCL-2 Proc Natl Acad Sci USA 1997 94: 11357–11362
Marzo I, Brenner C, Zamzami N, Jurgensmeier JM, Susin SA, Vieira HL, Prevost MC, Xie Z, Matsuyama S, Reed JC, Kroemer G . Bax and adenine nucleotide translocator cooperate in the mitochondrial control of apoptosis Science 1998 281: 2027–2031
Hu Y, Benedict MA, Ding L, Nunez, G . Role of cytochrome c and dATP/ATP hydrolysis in Apaf-1-mediated caspase-9 activation and apoptosis EMBO J 1999 18: 3586–3595
Li P, Nijhawan D, Budihardjo I, Srinivasula SM, Ahmad M, Alnemri ES, Wang X . Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade Cell 1997 91: 479–489
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Snow BE, Brothers GM, Mangion J, Jacotot E, Costantini P, Loeffler M, Larochette N, Goodlett DR, Aebersold R, Siderovski DP, Penninger JM, Kroemer G . Molecular characterization of mitochondrial apoptosis-inducing factor Nature 1999 397: 441–446
Susin SA, Zamzami N, Castedo M, Hirsch T, Marchetti P, Macho A, Daugas E, Geuskens M, Kroemer G . Bcl-2 inhibits the mitochondrial release of an apoptogenic protease J Exp Med 1996 184: 1331–1341
Yang J, Liu XS, Bhalla K, Kim CN, Ibrado AM, Cai JY, Peng TI, Jones DP, Wang XD . Prevention of apoptosis by Bcl-2. Release of cytochrome c from mitochondria blocked Science 1997 275: 1129–1132
Eskes R, Desagher S, Antonsson B, Martinou JC . Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane Mol Cell Biol 2000 20: 929–935
Pinkoski MJ, Green DR . Fas ligand, death gene Cell Death Differ 1999 6: 1174–1181
Chinnaiyan AM, O'Rourke K, Tewari M, Dixit VM . FADD, a novel death domain-containing protein, interacts with the death domain of Fas and initiates apoptosis Cell 1995 81: 505–512
Medema JP, Scaffidi C, Kischkel FC, ShevchenkonA, Mann M, Krammer PH, Peter ME . FLICE is activated by association with the CD95 death-inducing signaling complex (DISC) EMBO J 1997 16: 2794–2804
Scaffidi C, Schmitz I, Zha J, Korsmeyer SJ, Krammer PH, Peter ME . Differential modulation of apoptosis sensitivity in CD95 type I and type II cells J Biol Chem 1999 274: 22532–22538
Li H, Zhu H, Xu CJ, Yuan J . Cleavage of Bid by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis Cell 1998 94: 491–501
Luo X, Budihardjo I, Zou H, Slaughter C, Wang X . Bid, a Bcl2 interacting protein, mediates cytochrome c release from mitochondria in response to activation of cell surface death receptors Cell 1998 94: 481–490
Albanese J, Dainiak N . Ionizing radiation alters Fas antigen ligand at the cell surface and on exfoliated plasma membrane-derived vesicles: implications for apoptosis and intercellular signaling Radiat Res 2000 153: 49–61
Friesen C, Herr I, Krammer PH, Debatin KM . Involvement of the CD95 (APO-1/FAS) receptor/ligand system in drug-induced apoptosis in leukemia cells Nature Med 1996 2: 574–577
Fulda S, Strauss G, Meyer E, Debatin KM . Functional CD95 ligand and CD95 death-inducing signaling complex in activation-induced cell death and doxorubicin-induced apoptosis in leukemic T cells Blood 2000 95: 301–308
Muller M, Wilder S, Bannasch D, Israeli D, Lehlbach K, Li-Weber M, Friedman SL, Galle PR, Stremmel W, Oren M, Krammer PH . p53 activates the CD95 (APO-1/Fas) gene in response to DNA damage by anticancer drugs J Exp Med 1998 188: 2033–2045
Ogawa Y, Nishioka A, Hamada N, Terashima M, Inomata T, Yoshida S, Seguchi H, Kishimoto S . Expression of fas (CD95/APO-1) antigen induced by radiation therapy for diffuse B-cell lymphoma: immunohistochemical study Clin Cancer Res 1997 3: 2211–2216
Teitz T, Wei T, Valentine MB, Vanin EF, Grenet J, Valentine VA, Behm FG, Look AT, Lahti JM, Kidd VJ . Caspase 8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN Nature Med 2000 6: 529–535
Yeh WC, Pompa JL, McCurrach ME, Shu HB, Elia AJ, Shahinian A, Ng M, Wakeham A, Khoo W, Mitchell K, El-Deiry WS, Lowe SW, Goeddel DV, Mak TW . FADD: essential for embryo development and signaling from some, but not all, inducers of apoptosis Science 1998 279: 1954–1958
Varfolomeev EE, Schuchmann M, Luria V, Chiannilkulchai N, Beckmann JS, Mett IL, Rebrikov D, Brodianski VM, Kemper OC, Kollet O, Lapidot T, Soffer D, Sobe T, Avraham KB, Goncharov T, Holtmann H, Lonai P, Wallach D . Targeted disruption of the mouse Caspase 8 gene ablates cell death induction by the TNF receptors, Fas/Apo1, and DR3 and is lethal prenatally Immunity 1998 9: 267–276
Micheau O, Solary E, Hammann A, Dimanche-Boitrel MT . Fas ligand-independent, FADD-mediated activation of the Fas death pathway by anticancer drugs J Biol Chem 1999 274: 7987–7992
Rieux-Laucat F, Blachere S, Danielan S, de Villartay JP, Oleastro M, Solary E, Badre Meunier B, Arkwright P, Pondaré C, Bernaudin F, Chapel H, Nielsen S, Berah M, Fisher A, Le Deist F . Lymphoproliferative syndrome with autoimmunity: a possible genetic basis for dominant expression of the clinical manifestations Blood 1999 94: 1192–1199
Landowski TH, Qu N, Buyuksal I, Painter JS, Dalton WS . Mutations in the Fas antigen in patients with multiple myeloma Blood 1997 90: 4266–4270
Beltinger C, Kurz E, Bohler T, Schrappe M, Ludwig WD, Debatin KM . CD95 (APO-1/Fas) mutations in childhood T-lineage acute lymphoblastic leukemia Blood 1998 91: 3943–3951
Tamiya S, Etoh K, Suzushima H, Takatsuki K, Matsuoka M . Mutation of CD95 (Fas/Apo-1) gene in adult T cell leukemia cells Blood 1998 91: 3935–3942
Maeda T, Yamada Y, Moriuchi R, Sugahara K, Tsuruda K, Joh T, Atogami S, Tsukasaki K, Tomonaga M, Kamihira S . Fas gene mutation in the progression of adult T cell leukemia J Exp Med 1999 189: 1063–1071
Wang J, Zheng L, Lobito A, Chan FK, Dale J, Sneller M, Yao X, Puck JM, Straus SE, Lenardo MJ . Inherited human caspase 10 mutations underlie defective lymphocyte and dendritic cell apoptosis in autoimmune lymphoproliferative syndrome type II Cell 1999 98: 47–58
Budihardjo I, Oliver H, Lutter M, Luo X, Wang X . Biochemical pathways of caspase activation during apoptosis Annu Rev Cell Dev Biol 1999 15: 269–290
Nicholson DW . Caspase structure, proteolytic substrates, and function during apoptotic cell death Cell Death Differ 1999 6: 1028–1042
Walker NP, Talanian RV, Brady KD, Dang LC, Bump NJ, Ferenz CR, Franklin S, Ghayur T, Hackett MC, Hammill LD, Herzog L, Hugunin M, Houy W, Mankovich JA, McGuiness L, Orlewicz E, Paskind M, Pratt CA, Reis P, Summani A, Terranova M, Welch JP, Xiong L, Möller A, Tracey DE, Kamen R, Wong WW . Crystal structure of the cysteine protease interleukin-1 beta-converting enzyme: a (p20/p10)2 homodimer Cell 1994 78: 343–352
Rotonda J, Nicholson DW, Fazil KM, Gallant M, Gareau Y, Labelle M, Peterson EP, Rasper DM, Ruel R, Vaillancourt JP, Thornberry NA, Becker JW . The three-dimensional structure of apopain/CPP32, a key mediator of apoptosis Nat Struct Biol 1996 3: 619–625
Zou H, Henzel WJ, Liu X, Lutschg A, Wang X . Apaf-1, a human protein homologous to C. elegans CED-4, participates in cytochrome c-dependent activation of caspase-3 Cell 1997 90: 405–413
Duan H, Dixit VM . RAIDD is a new ‘death’ adaptor molecule Nature 1997 385: 86–89
Ahmad M, Srinivasula SM, Wang L . CRADD, a novel human apoptotic adaptor molecule for caspase-2, and FasL/tumor necrosis factor receptor-interacting protein RIP Cancer Res 1997 57: 615–619
Slee EA, Harte MT, Kluck RM, Wolf BB, Casiano CA, Newmeyer DD, Wang HG, Reed JC, Nicholson DW, Alnemri ES, Green DR, Martin SJ . Ordering the cytochrome c-initiated caspase cascade: hierarchical activation of caspases-2, -3, -6, -7, -8, and -10 in a caspase-9-dependent manner J Cell Biol 1999 144: 281–292
Dubrez L, Savoy I, Hamman A, Solary E . Pivotal role of a DEVD-sensitive step in etoposide-induced and Fas-mediated apoptotic pathways EMBO J 1996 15: 5504–5512
Dubrez L, Eymin B, Sordet O, Droin N, Turhan AG, Solary E . BCR-ABL delays apoptosis upstream of procaspase-3 activation Blood 1998 91: 2415–2422
Droin N, Dubrez L, Eymin B, Renvoize C, Breard J, Dimanche-Boitrel MT, Solary E . Upregulation of CASP genes in human tumor cells undergoing etoposide-induced apoptosis Oncogene 1998 16: 2885–2894
Sordet O, Bettaieb A, Bruey JM, Eymin B, Droin N, Ivarsson M, Garrido C, Solary E . Selective inhibition of apoptosis by TPA-induced differentiation of U937 leukemic cells Cell Death Differ 1999 6: 351–361
Kuida K, Haydar TF, Kuan CY, Gu Y, Taya C, Karasuyama H, Su MS, Rakic P, Flavell RA . Reduced apoptosis and cytochrome c-mediated caspase activation in mice lacking caspase-9 Cell 1998 94: 325–337
Hakem R, Hakem A, Duncan GS, Henderson JT, Woo M, Soengas MS, Elia A, de la Pompa JL, Kagi D, Khoo W, Potter J, Yoshida R, Kaufman SA, Lowe SW, Penninger JM, Mak TW . Differential requirement for caspase-9 in apoptotic pathways in vivo Cell 1998 94: 339–352
Cecconi F, Alvarez-Bolado G, Meyer BI, Roth KA, Gruss P . Apaf1 (CED-4 homolog) regulates programmed cell death in mammalian development Cell 1998 94: 727–737
Yoshida H, Kong YY, Yoshida R, Elia AJ, Hakem A, Hakem R, Penninger JM, Mak TW . Apaf1 is required for mitochondrial pathways of apoptosis and brain development Cell 1998 94: 739–750
Sakahira H, Enari M, Nagata S . Cleavage of CAD inhibitor in CAD activation and DNA degradation during apoptosis Nature 1998 391: 96–99
Sahara S, Aoto M, Eguchi Y, Imamoto N, Yoneda Y, Tsujimoto Y . Acinus is a caspase-3-activated protein required for apoptotic chromatin condensation Nature 1999 401: 168–173
Samali A, Cai J, Zhivotovsky B, Jones DP, Orrenius S . Presence of a pre-apoptotic complex of pro-caspase-3, Hsp60 and Hsp10 in the mitochondrial fraction of jurkat cells EMBO J 1999 18: 2040–2048
Xanthoudakis S, Roy S, Rasper D, Hennessay T, Aubin Y, Cassady R, Tawa P, Ruel R, Rosen A, Nicholson DW . Hsp60 accelerates the maturation of pro-caspase-3 by upstream activator proteases during apoptosis EMBO J 1999 18: 2049–2056
Susin SA, Lorenzo HK, Zamzami N, Marzo I, Brenner C, Larochette N, Prevost MC, Alzari PM, Kroemer G . Mitochondrial release of caspase-2 and-9 during the apoptotic process J Exp Med 1999 189: 381–394
Krajewski S, Krajewska M, Ellerby LM, Welsh K, Xie Z, Deveraux QL, Salvesen GS, Bredesen DE, Rosenthal RE, Fiskum G, Reed JC . Release of caspase-9 from mitochondria during neuronal apoptosis and cerebral ischemia Proc Natl Acad Sci USA 1999 96: 5752–5757
Chandler JM, Cohen GM, MacFarlane M . Different subcellular distribution of caspase-3 and caspase-7 following Fas-induced apoptosis in mouse liver J Biol Chem 1998 273: 10815–10818
Kumar A, Commane M, Flickinger TW, Horvath CM, Stark GR . Defective TNFa-induced apoptosis in STAT1-null cells due to low constitutive levels of caspases Science 1997 278: 1630–1632
Micheau O, Hammann A, Solary E, Dimanche-Boitrel MT . STAT-1-independent upregulation of FADD and procaspase-3 and -8 in cancer cells treated with cytotoxic drugs Biochem Biophys Res Commun 1999 256: 603–607
Wang L, Miura M, Bergeron L, Zhu H, Yuan J . Ich-1, an Ice/ced-3-related gene, encodes both positive and negative regulators of programmed cell death Cell 1994 78: 739–750
Srinivasula SM, Ahmad M, Guo Y, Zhan Y, Lazebnik Y, Fernandes-Alnemri T, Alnemri ES . Identification of an endogenous dominant-negative short isoform of caspase-9 that can regulate apoptosis Cancer Res 1999 59: 999–1002
Seol DW, Billiar TR . A caspase-9 variant missing the catalytic site is an endogenous inhibitor of apoptosis J Biol Chem 1999 274: 2072–2076
Dimmeler S, Haendeler J, Nehls M, Zeiher AM . Suppression of apoptosis by nitric oxide via inhibition of interleukin-1beta-converting enzyme (ICE)-like and cysteine protease protein (CPP)-32-like proteases J Exp Med 1997 185: 601–607
Mannick JB, Hausladen A, Liu L, Hess DT, Zeng M, Miao QX, Kane LS, Gow AJ, Stamler JS . Fas-induced caspase denitrosylation Science 1999 284: 651–654
Cardone MH, Roy N, Stennicke HR, Salvesen GS, Franke TF, Stanbridge E, Frisch S, Reed JC . Regulation of cell death protease caspase-9 by phosphorylation Science 1998 282: 1318–1321
Schurmann A, Mooney AF, Sanders LC, Sells MA, Wang HG, Reed JC, Bokoch GM . p21-activated kinase 1 phosphorylates the death agonist Bad and protects cells from apoptosis Mol Cell Biol 2000 20: 453–461
Deveraux QL, Stennicke HR, Salvesen GS, Reed JC . Endogenous inhibitors of caspases J Clin Immunol 1999 19: 388–398
Ambrosini G, Adida C, Altieri DC . A novel anti-apoptosis gene, survivin, expressed in cancer and lymphoma Nature Med 1997 3: 917–921
Tamm I, Wang Y, Sausville E, Scudiero DA, Vigna N, Oltersdorf T, Reed JC . IAP-family protein survivin inhibits caspase activity and apoptosis induced by Fas (CD95), Bax, caspases, and anticancer drugs Cancer Res 1998 58: 5315–5320
Ambrosini G, Adida C, Sirugo G, Altieri DC . Induction of apoptosis and inhibition of cell proliferation by survivin gene targeting J Biol Chem 1998 273: 11177–11182
Kobayashi K, Hatano M, Otaki M, Ogasawara T, Tokuhisa T . Expression of a murine homologue of the inhibitor of apoptosis protein is related to cell proliferation Proc Natl Acad Sci USA 1999 96: 1457–1462
Li F, Ambrosini G, Chu EY, Plescia J, Tognin S, Marchisio PC, Altieri DC . Control of apoptosis and mitotic spindle checkpoint by survivin Nature 1998 396: 580–584
Li F, Ackermann EJ, Bennett CF, Rothermel AL, Plescia J, Tognin S, Villa A, Marchisio PC, Altieri DC . Pleiotropic cell-division defects and apoptosis induced by interference with survivin function Nat Cell Biol 1999 1: 461–466
Tschopp J, Irmler M, Thome M . Inhibition of Fas death signals by FLIPs Curr Opin Immunol 1998 10: 552–558
Bakhshi A, Jensen JP, Goldman P, Wright JJ, McBride OW, Epstein AL, Korsmeyer SJ . Cloning the chromosomal breakpoint of t(14;18) human lymphomas: clustering around JH on chromosome 14 and near a transcriptional unit on 18 Cell 1985 41: 899–906
Cleary ML, Smith SD, Sklar J . Cloning and structural analysis of cDNAs for bcl-2 and a hybrid bcl-2/immunoglobulin transcript resulting from the t(14;18) translocation Cell 1986 47: 19–28
Zamzami N, Brenner C, Marzo I, Susin SA, Kroemer G . Subcellular and submitochondrial mode of action of Bcl-2-like oncoproteins Oncogene 1998 16: 2265–2282
Gross A, McDonnell JM, Korsmeyer SJ . BCL-2 family members and the mitochondria in apoptosis Genes Dev 1999 13: 1899–1911
Strasser A, Harris AW, Bath ML, Cory S . Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2 Nature 1990 348: 331–333
McDonnell TJ, Deane N, Platt FM, Nunez G, Jaeger U, McKearn JP, Korsmeyer SJ . bcl-2-immunoglobulin transgenic mice demonstrate extended B cell survival and follicular lymphoproliferation Cell 1989 57: 79–88
McDonnell TJ, Korsmeyer SJ . Progression from lymphoid hyperplasia to high-grade malignant lymphoma in mice transgenic for the t(14;18) Nature 1991 349: 254–256
Meijerink JP, Mensink EJ, Wang K, Sedlak TW, Sloetjes AW, de Witte T, Waksman G, Korsmeyer SJ . Hematopoietic malignancies demonstrate loss-of-function mutations of BAX Blood 1998 91: 2991–2997
Reed JC . Bcl-2 family proteins: relative importance as determinants of chemoresistance in cancer. In: Hickman JA, Dive C (eds) Apoptosis and Cancer Chemotherapy Humana Press: Totowa, NJ 1999 99–116
Kornblau SM, Thall PF, Estrov Z, Walterscheid M, Patel S, Theriault A, Keating MJ, Kantarjian H, Estey E, Andreeff M . The prognostic impact of BCL2 protein expression in acute myelogenous leukemia varies with cytogenetics Clin Cancer Res 1999 5: 1758–1766
Kaufmann SH, Karp JE, Svingen PA, Krajewski S, Burke PJ, Gore SD, Reed JC . Elevated expression of the apoptotic regulator Mcl-1 at the time of leukemic relapse Blood 1998 91: 991–1000
Hogarth LA, Hall AG . Increased BAX expression is associated with an increased risk of relapse in childhood acute lymphocytic leukemia Blood 1999 93: 2671–2678
Houghton JA . Apoptosis and drug response Curr Opin Oncol 1999 11: 475–481
Vander Heiden MG, Thompson CB . Bcl-2 proteins: regulators of apoptosis or of mitochondrial homeostasis? Nature Cell Biol 1999 1: E209–E216
Antonsson B, Conti F, Ciavatta A, Montessuit S, Lewis S, Martinou I, Bernasconi L, Bernard A, Mermod JJ, Mazzei G, Maundrell K, Gambale F, Sadoul R, Martinou JC . Inhibition of Bax channel-forming activity by Bcl-2 Science 1997 277: 370–372
Shibasaki F, Kondo E, Akagi T, McKeon F . Suppression of signalling through transcription factor NF-AT by interactions between calcineurin and Bcl-2 Nature 1997 386: 728–731
Miyashita T, Reed JC . Bcl-2 oncoproteins blocks chemotherapy-induced apoptosis in a human leukemia cell line Blood 1993 81: 151–157
Schmitt E, Cimoli G, Steyaert A, Bertrand R . Bcl-xL modulates apoptosis induced by anticancer drugs and delays DEVDase and DNA fragmentation-promoting activities Exp Cell Res 1998 240: 107–121
Kitada S, Takayama S, De Riel K, Tanaka S, Reed JC . Reversal of chemoresistance of lymphoma cells by antisense-mediated reduction of bcl-2-gene expression Antisense Res Dev 1994 4: 71–79
Jansen B, Schlagbauer-Wald H, Brown BD, Bryan RN, Van Elsas A, Müller M, Wolff K, Eichler H-G, Pehamberger H . Bcl-2 antisense therapy chemosensitizes human melanoma in SCID mice Nature Med 1998 4: 232–234
Tai YT, Strobel T, Kufe D, Cannistra SA . In vivo cytotoxicity of ovarian cancer cells through tumor-selective expression of the BAX gene Cancer Res 1999 59: 2121–2126
Haldar S, Jena N, Croce CM . Inactivation of Bcl-2 by phosphorylation Proc Natl Acad Sci USA 1995 92: 4507–4511
Blagosklonny MV, Schulte T, Phuongmai N, Trepel J, Neckers LM . Taxol-induced apoptosis and phosphorylation of Bcl-2 protein involves c-Raf-1 and represents a novel c-Raf-1 signal tranduction pathway Cancer Res 1996 56: 1851–1854
Basu A, Haldar S . Microtubule-damaging drugs triggered Bcl2 phosphorylation-requirement of phosphorylation on both serine-70 and serine-87 residues of bcl2 protein Int J Oncol 1998 13: 659–664
Blagosklonny MV, Giannakakou P, El-Deiry W, Kingston DGI, Higgs PI, Neckers L, Fojo T . Raf-1/bcl-2 phosphorylation: a step from microtubule damage to cell death Cancer Res 1997 57: 130–135
Wang LG, Liu XM, Kreis W, Budman DR . The effect of antimicrotubule agents on signal transduction pathways of apoptosis Cancer Chemother Pharmacol 1999 44: 355–361
Srivastava RK, Srivastava AR, Korsmeyer SJ, Nesterova M, Cho-Chung YS, Longo DL . Involvement of microtubules in the regulation of Bcl2 phosphorylation and apoptosis through cyclic AMP-dependent protein kinase Mol Cell Biol 1998 18: 3509–3517
Blagosklonny MV, Chuman Y, Bergan RC, Fojo T . Mitogen-activated protein kinase pathway is dispensable for microtubule-active drug-induced Raf-1/Bcl-2 phosphorylation and apoptosis in leukemia cells Leukemia 1999 13: 1028–1036
Basu A, You SA, Haldar S . Regulation of Bcl2 phosphorylation by stress response kinase pathway Int J Oncol 2000 16: 497–500
Attala H, Westberg JA, Andersson LC, Adlercreutz H, Makela TP . 2-Methyoxyestradiol-induced phosphorylation of bcl-2: uncoupling from JNK/SAPK activation Biochem Biophys Res Commun 1998 247: 616–621
Nagata S . Biddable death Nat Cell Biol 1999 1: E143–E145
Desagher S, Osen-Sand A, Nichols A, Eskes R, Montessuit S, Lauper S, Maundrell K, Antonsson B, Martinou JC . Bid-induced conformational change of Bax is responsible for mitochondrial cytochrome c release during apoptosis J Cell Biol 1999 144: 891–901
Downward J . How BAD phosphorylation is good for survival Nat Cell Biol 1999 1: E33–E35
Wang HG, Pathan N, Ethell IM, Krajewski S, Yamaguchi Y, Shibasaki F, McKeon F, Bobo T, Franke TF, Reed JC . Ca2+-induced apoptosis through calcineurin dephosphorylation of BAD Science 1999 284: 339–343
Puthalakath H, Huang DC, O'Reilly LA, King SM, Strasser A . The proapoptotic activity of the Bcl-2 family member Bim is regulated by interaction with the dynein motor complex Mol Cell 1999 3: 287–296
Bouillet P, Metcalf D, Huang DC, Tarlinton DM, Kay TW, Kontgen F, Adams JM, Strasser A . Proapoptotic Bcl-2 relative Bim required for certain apoptotic responses, leukocyte homeostasis, and to preclude autoimmunity Science 1999 286: 1735–1738
Cheng EH, Kirsch DG, Clem RJ, Ravi R, Kastan MB, Bedi A, Ueno K, Hardwick JM . Conversion of Bcl-2 to a Bax-like death effector by caspases Science 1997 278: 1966–1968
Selvakumaran M, Lin HK, Miyashita T, Wang HG, Krajewski S, Reed JC, Hoffman B, Liebermann D . Immediate early up-regulation of bax expression by p53 but not TGF beta 1: a paradigm for distinct apoptotic pathways Oncogene 1994 9: 1791–1798
Basu A, Haldar S . The relationship between Bcl-2, Bax and p53: consequences for cell cycle progression and cell death Mol Hum Reprod 1998 4: 1099–1109
Inohara N, Ding L, Chen S, Nunez G . Harakiri, a novel regulator of cell death, encodes a protein that activates apoptosis and interacts selectively with survival-promoting proteins Bcl-2 and Bcl-X(L) EMBO J 1997 16: 1686–1694
Jaattela M . Heat shock proteins as cellular lifeguards Ann Med 1999 31: 261–271
Samali A, Holmberg CI, Sistonen L, Orrenius S . Thermotolerance and cell death are distinct cellular responses to stress: dependence on heat shock proteins FEBS Lett 1999 461: 306–310
Galea-Lauri J, Richardson AJ, Latchman DS, Katz DR . Increased heat shock protein 90 (hsp90) expression leads to increased apoptosis in the monoblastoid cell line U937 following induction with TNF-alpha and cycloheximide: a possible role in immunopathology J Immunol 1996 157: 4109–4118
Jaattela M, Wissing D, Kokholm K, Kallunki T, Egeblad M . Hsp70 exerts its anti-apoptotic function downstream of caspase-3-like proteases EMBO J 1998 17: 6124–6134
Stuart JK, Myszka DG, Joss L, Mitchell RS, McDonald SM, Xie Z, Takayama S, Reed JC, Ely KR . Characterization of interactions between the anti-apoptotic protein BAG-1 and Hsc70 molecular chaperones J Biol Chem 1998 273: 22506–22514
Gabai VL, Meriin AB, Mosser DD, Caron AW, Rits S, Shifrin VI, Sherman MY . Hsp70 prevents activation of stress kinases. A novel pathway of cellular thermotolerance J Biol Chem 1997 272: 18033–18037
Mosser DD, Caron AW, Bourget L, Denis-Larose C, Massie B . Role of the human heat shock protein hsp70 in protection against stress-induced apoptosis Mol Cell Biol 1997 17: 5317–5327
Buzzard KA, Giaccia AJ, Killender M, Anderson RL . Heat shock protein 72 modulates pathways of stress-induced apoptosis J Biol Chem 1998 273: 17147–17153
Garrido C, Fromentin A, Bonnotte B, Favre N, Moutet M, Arrigo AP, Mehlen P, Solary E . Heat shock protein 27 enhances the tumorigenicity of immunogenic rat colon carcinoma cell clones Cancer Res 1998 58: 5495–5499
Mehlen P, Schulze-Osthoff K, Arrigo AP . Small stress proteins as novel regulators of apoptosis. Heat shock protein 27 blocks Fas/APO-1- and staurosporine-induced cell death J Biol Chem 1996 271: 16510–16514
Garrido C, Ottavi P, Fromentin A, Hammann A, Arrigo AP, Chauffert B, Mehlen P . HSP27 as a mediator of confluence-dependent resistance to cell death induced by anticancer drugs Cancer Res 1997 57: 2661–2667
Mehlen P, Kretz-Remy C, Preville X, Arrigo AP . Human hsp27, Drosophila hsp27 and human alphaB-crystallin expression-mediated increase in glutathione is essential for the protective activity of these proteins against TNFalpha-induced cell death EMBO J 1996 15: 2695–2706
Lavoie JN, Lambert H, Hickey E, Weber LA, Landry J . Modulation of cellular thermoresistance and actin filament stability accompanies phosphorylation-induced changes in the oligomeric structure of heat shock protein 27 Mol Cell Biol 1995 15: 505–516
Konishi H, Matsuzaki H, Tanaka M, Takemura Y, Kuroda S, Ono Y, Kikkawa U . Activation of protein kinase B (Akt/RAC-protein kinase) by cellular stress and its association with heat shock protein Hsp27 FEBS Lett 1997 410: 493–498
Garrido C, Bruey JM, Fromentin A, Hammann A, Arrigo AP, Solary E . HSP27 inhibits cytochrome c-dependent activation of procaspase-9 FASEB J 1999 13: 2061–2070
Bruey JM, Ducasse C, Bonniaud P, Ravagnan L, Susin SA, Diaz-Latoud C, Gurbuxani S, Arrigo AP, Kroemer G, Solary E, Garrido C . Hsp27 negatively regulates cell death by interacting with cytochrome c Nature Cell Biol 2000 (in press
Kim SH, Kim D, Han JS, Jeong CS, Chung BS, Kang CD, Li GC . Ku autoantigen affects the susceptibility to anticancer drugs Cancer Res 1999 59: 4012–4017
Martinez-Lorenzo MJ, Gamen S, Etxeberria J, Lasierra P, Larrad L, Pineiro A, Anel A, Naval J, Alava MA . Resistance to apoptosis correlates with a highly proliferative phenotype and loss of Fas and CPP32 (caspase-3) expression in human leukemia cells Int J Cancer 1998 75: 473–481
von Reyher U, Strater J, Kittstein W, Gschwendt M, Krammer PH, Moller P . Colon carcinoma cells use different mechanisms to escape CD95-mediated apoptosis Cancer Res 1998 58: 526–534
Algeciras-Schimnich A, Griffith TS, Lynch DH, Paya CV . Cell cycle-dependent regulation of FLIP levels and susceptibility to Fas-mediated apoptosis J Immunol 1999 162: 5205–5211
Micheau O, Solary E, Hammann A, Martin F, Dimanche-Boitrel MT . Sensitization of cancer cells treated with cytotoxic drugs to fas-mediated cytotoxicity J Natl Cancer Inst 1997 89: 783–789
Sheard MA, Krammer PH, Zaloudik J . Fractionated gamma-irradiation renders tumour cells more responsive to apoptotic signals through CD95 Br J Cancer 1999 80: 1689–1696
Posovszky C, Friesen C, Herr I, Debatin KM . Chemotherapeutic drugs sensitize pre-B ALL cells for CD95- and cytotoxic T-lymphocyte-mediated apoptosis Leukemia 1999 13: 400–409
Morimoto H, Yonehara S, Bonavida B . Overcoming tumor necrosis factor and drug resistance of human tumor cell lines by combination treatment with anti-Fas antibody and drugs or toxins Cancer Res 1993 53: 2591–2596
Lee KL, Spielmann J, Zhao DJ, Olsen KJ, Podack ER . Perforin, Fas ligand, and tumor necrosis factor are the major cytotoxic molecules used by lymphokine-activated killer cells J Immunol 1996 157: 1919–1925
Knight CRL, Rees RC, Platts A, Johnson T, Griffin M . Interleukin-2-activated human effector lymphocytes mediate cytotoxicity by inducing apoptosis in human leukaemia and solid tumour target cells Immunology 1993 79: 535–541
Kondo T, Suda T, Fukuyama H, Adachi M, Nagata S . Essential roles of the Fas ligand in the development of hepatitis Nature Med 1997 3: 409–413
Wiley SR, Schooley K, Smolak PJ, Din WS, Huang CP, Nicholl JK, Sutherland GR, Smith TD, Rauch C, Smith CA, Goodwin RG . Identification and characterization of a new member of the TNF family that induces apoptosis Immunity 1995 3: 673–682
Pitti RM, Marters SA, Ruppert TS, Donahue CJ, Moore A, Ashkenazi A . Induction of apoptosis by Apo-2 ligand, a new member of the tumor necrosis factor family J Biol Chem 1996 271: 12687–12690
Bodmer JL, Holler N, Reynard S, Vinciguerra P, Schneider P, Juo P, Blenis J, Tschopp J . TRAIL receptor-2 signals apoptosis through FADD and caspase-8 Nat Cell Biol 2000 2: 241–243
Walczak H, Miller RE, Ariail K, Gliniak B, Griffith TS, Kubin M, Chin W, Jones J, Woodward A, Le T, Smith C, Smolak P, Goodwin RG, Rauch CT, Schuh JC, Lynch DH . Tumoricidal activity of tumor necrosis factor-related apoptosis-inducing ligand in vivo Nature Med 1999 5: 157–163
Ashkenazi A, Pai RC, Fong S, Leung S, Lawrence DA, Marsters SA, Blackie C, Chang L, McMurtrey AE, Hebert A, DeForge L, Koumenis IL, Lewis D, Harris L, Bussiere J, Koeppen H, Shahrokh Z, Schwall RH . Safety and antitumor activity of recombinant soluble Apo2 ligand J Clin Invest 1999 104: 155–162
Jo M, Kim TH, Seol DW, Esplen JE, Dorko K, Billiar TR, Strom SC . Apoptosis induced in normal human hepatocytes by tumor necrosis factor-related apoptosis inducing ligand Nature Med 2000 6: 564–567
Griffith TS, Lynch, DH . TRAIL: a molecule with multiple receptors and control mechanisms Curr Opin Immunol 1998 10: 559–563
Ashkenazi A, Dixit VM . Apoptosis control by death and decoy receptors Curr Opin Cell Biol 1999 11: 255–260
Mizutani Y, Yoshida O, Miki T, Bonavida B . Synergistic cytotoxicity and apoptosis by Apo-2 ligand and adriamycin against bladder cancer cells Clin Cancer Res 1999 5: 2605–2612
Bonavida B, Ng CP, Jazirehi A, Schiller G, Mizutani Y . Selectivity of TRAIL-mediated apoptosis of cancer cells and synergy with drugs: the trail to non-toxic cancer therapeutics Int J Oncol 1999 15: 793–802
Gibson SB, Oyer R, Spalding AC, Anderson SM, Johnson GL . Increased expression of death receptors 4 and 5 synergizes the apoptosis response to combined treatment with etoposide and TRAIL Mol Cell Biol 2000 20: 205–212
Zhang XD, Franco A, Myers K, Gray C, Nguyen T, Hersey P . Relation of TNF-related apoptosis-inducing ligand (TRAIL) receptor and FLICE-inhibitory protein expression to TRAIL-induced apoptosis of melanoma Cancer Res 1999 59: 2747–2753
Woo RA, McLure KG, Lees-Miller SP, Rancourt DE, Lee PMK . DNA-dependent kinase acts upstream of p53 in response to DNA damage Nature 1998 394: 700–704
Waterman MJ, Stavridi ES, Waterman JL, Halazonetis TD . ATM-dependent activation of p53 involves dephosphorylation and association with 14-3-3 proteins Nat Genet 1998 19: 175–178
White E, Prives C . DNA damage enables p73 Nature 1999 399: 734–535
Gong JG, Costanzo A, Yang HQ, Melino G, Kaelin WG Jr, Levrero M, Wang JY . The tyrosine kinase c-Abl regulates p73 in apoptotic response to cisplatin-induced DNA damage Nature 1999 399: 806–809
Agami R, Blandino G, Oren M, Shaul Y . Interaction of c-Abl and p73 alpha and their collaboration to induce apoptosis Nature 1999 399: 809–813
Yuan ZM, Shioya H, Ishiko T, Sun X, Gu J, Huang YY, Lu H, Kharbanda S, Weichselbaum R, Kufe D . p73 is regulated by tyrosine kinase c-Abl in the apoptotic response to DNA damage Nature 1999 399: 814–817
Miyashita T, Harigal M, Hanada M, Reed JC . Identification of p53-dependent negative responsive element in the bcl-2 gene Cancer Res 1994 54: 3131–3135
Miyashita T, Reed JC . Tumor suppressor p53 is a direct transcriptional activator of the human bax gene Cell 1995 80: 293–299
Tanaka H, Arakawa H, Yamaguchi T, Shiraishi K, Fukuda S, Matsui K, Takei Y, Nakamura Y . A ribonucleotide reductase gene involved in a p53-dependent cell-cycle checkpoint for DNA damage Nature 2000 404: 42–49
Sugiyama K, Shimizu M, Akiyama T, Tamaoki T, Yamaguchi K, Takahashi R, Eastman A, Akinaga S . UCN-01 selectively enhances mitomycin C cytotoxicity in p53 defective cells which is mediated through S and/or G(2) checkpoint abrogation Int J Cancer 2000 85: 703–709
Chan TA, Hermeking H, Lengauer C, Kinzler KW, Vogelstein B . 14–3–3σ is required to prevent mitotic catastrophe after DNA damage Nature 1999 401: 616–620
Lowe SW, Ruley HE, Jacks T, Housman DE . p53-dependent apoptosis modulates the cytotoxicity of anticancer agents Cell 1993 74: 957–967
Lowe SW, Bodis S, McClatchey A, Remington L, Ruley HE, Fisher DE, Housman DE, Jacks T . p53 status and the efficacy of cancer therapy in vivo Science 1994 266: 807–810
Weinstein JN, Myers TG, O'Connor PM, Friend SH, Fornace AJ Jr, Kohn KW, Fojo T, Bates SE, Rubinstein LV, Anderson NL, Buolamwini JK, van Osdol WW, Monks AP, Scudiero DA, Sausville EA, Zaharevitz DW, Bunow B, Viswanadhan VN, Johnson GS, Wittes RE, Paull KD . An information-intensive approach to the molecular pharmacology of cancer Science 1997 275: 343–349
Waldman T, Zhang Y, Dillehay L, Yu J, Kinzler K, Vogelstein B, Williams J . Cell-cycle arrest versus cell death in cancer therapy Nature Med 1997 3: 1034–1036
Zindy F, Eischen CM, Randle DH, Kamijo T, Cleveland JL, Sherr CJ, Roussel MF . Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization Genes Dev 1998 12: 2424–2433
Bissonnette RP, Echeverri F, Mahboubi A, Green DR . Apoptotic cell death induced by c-myc is inhibited by bcl-2 Nature 1992 359: 552–554
Brown JM, Wouters BG . Apoptosis, p53 and tumor cell sensitivity to anticancer agents Cancer Res 1999 59: 1391–1399
Komarov PG, Komarova EA, Kondratov RV, Christov-Tselkov K, Coon JS, Chernov MV, Gudkov AV . A chemical inhibitor of p53 that protects mice from the side-effects of cancer therapy Science 1999 285: 1733–1737
Rayet B, Gelinas C . Aberrant rel/NF-κB genes and activity in human cancer Oncogene 1999 18: 6938–6947
Beg AA, Baltimore D . An essential role for NF-κB in preventing TNF-a-induced apoptosis by NF-κB Science 1996 274: 787–789
Wang CY, Mayo MW, Baldwin AS Jr . TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB Science 1996 274: 784–787
Furman RR, Asgary Z, Mascarenhas JO, Liou HC, Schattner EJ . Modulation of NF-Kappa B activity and apoptosis in chronic lymphocytic leukemia B cells J Immunol 2000 164: 2200–2206
Kordes U, Krappmann D, Heissmeyer V, Ludwig WD, Scheidereit C . Transcription factor NF-kappaB is constitutively activated in acute lymphoblastic leukemia cells Leukemia 2000 14: 399–402
Luque I, Gelinas C . Rel/NF-κB and I-κB factors in oncogenesis Semin Cancer Biol 1997 8: 103–111
LaCasse EC, Baird S, Korneluk RG, MacKenzie AE . The inhibitors of apoptosis (IAPs) and their emerging role in cancer Oncogene 1998 17: 3247–3259
Chu ZL, McKinsey TA, Liu L, Gentry JJ, Malim MH, Ballard DW . Suppression of tumor necrosis factor-induced cell death by inhibitor of apoptosis c-iap2 is under NF-κB control Proc Natl Acad Sci USA 1997 94: 10057–10062
Stehlik C, de Martin R, Kumabashiri I, Schmid JA, Binder BR, Lipp J . Nuclear factor (NF)-κB-regulated X-chromosome-linked iap gene expression protects endothelial cells from tumor necrosis factor-a-induced apoptosis J Exp Med 1998 188: 211–216
Bargou RC, Emmerich F, Krappmann D, Bommert K, Mapara MY, Arnold W, Royer HD, Grinstein E, Greiner A, Scheidereit C, Dorken B . Constitutive nuclear factor-κB-RelA activation is required for proliferation and survival of Hodgkin's disease tumor cells J Clin Invest 1997 100: 2961–2969
Messineo C, Jamerson MH, Hunter E, Braziel R, Bagg A, Irving SG, Cossman J . Gene expression by single Reed–Sternberg cells: pathways of apoptosis and activation Blood 1998 91: 2443–2451
Kuhnel F, Zender L, Paul Y, Tietze MK, Trautwein C, Manns M, Kubicka S . NKkappaB mediated apoptosis through transcriptional activation of Fas (CD95) in adenoviral hepatitis J Biol Chem 2000 275: 6421–6427
Quignon F, De Bels F, Koken M, Feunteun J, Ameisen JC, de Thé H . PML induces a novel caspase-independent death process Nat Genet 1998 20: 259–265
Wang ZG, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R, Pandolfi PP . PML is essential for multiple apoptotic pathways Nat Genet 1998 20: 266–272
Nason-Burchenal K, Gandini D, Botto M, Allopenna J, Seale JR, Cross NC, Goldman JM, Dmitrovsky E, Pandolfi PP . Interferon augments PML and PML/RAR alpha expression in normal myeloid and acute promyelocytic cells and cooperates with all-trans retinoic acid to induce maturation of a retinoid-resistant promyelocytic cell line Blood 1996 88: 3926–3936
Dyck JA, Maul GG, Miller WH Jr, Chen JD, Kakizuka A, Evans RM . A novel macromolecular structure is a target of the promyelocyte-retinoic acid receptor oncoprotein Cell 1994 76: 333–343
Hodges M, Tissot C, Howe K, Grinwade D, Freemont PS . Structure, organization, and dynamics of promyelocytic leukemia protein nuclear bodies Am J Hum Genet 1998 63: 297–304
He LZ, Guidez F, Tribioli C, Peruzzi D, Ruthardt M, Zelent A, Pandolfi PP . Distinct interactions of PML-RARalpha and PLZF-RARalpha with co-repressors determine differential responses to RA in APL Nat Genet 1998 18: 126–135
Koken MH, Puvion-Dutilleul F, Guillemin MC, Viron A, Linares-Cruz G, Stuurman N, de Jong L, Szostecki C, Calvo F, Chomienne C et al. The t(15;17) translocation alters a nuclear body in a retinoic acid-reversible fashion EMBO J 1994 13: 1073–1083
Grignani F, De Matteis S, Nervi C, Tomassoni L, Gelmetti V, Cioce M, Fanelli M, Ruthardt M, Ferrara FF, Zamir I, Seiser C, Grignani F, Lazar MA, Minucci S, Pelicci PG . Fusion proteins of the retinoic acid receptor-alpha recruit histone deacetylase in promyelocytic leukaemia Nature 1998 391: 815–818
Lin RJ, Nagy L, Inoue S, Shao W, Miller WH Jr, Evans RM . Role of the histone deacetylase complex in acute promyelocytic leukaemia Nature 1998 391: 811–814
Sternsdorf T, Puccetti E, Jensen K, Hoelzer D, Will H, Ottmann OG, Ruthardt M . PIC-1/SUMO-1-modified PML-retinoic acid receptor alpha mediates arsenic trioxide-induced apoptosis in acute promyelocytic leukemia Mol Cell Biol 1999 19: 5170–5178
Nervi C, Ferrara FF, Fanelli M, Rippo MR, Tomassini B, Ferrucci PF, Ruthardt M, Gelmetti V, Gambacorti-Passerini C, Diverio D, Grignani F, Pelicci PG, Testi R . Caspases mediate retinoic acid-induced degradation of the acute promyelocytic leukemia PML/RARalpha fusion protein Blood 1998 92: 2244–2251
Zu J, Koken MH, Quignon F, Chelbi-Alix MK, Degos L, Wang ZY, Chen Z, de Thé H . Arsenic-induced PML targeting on to nuclear bodies: implications for the treatment of acute promyelocytic leukemia Proc Natl Acad Sci USA 1997 94: 3978–3983
Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C, Grosveld F, Pandolfi PP . Role of PML in cell growth and the retinoic acid pathway Science 1998 279: 1547–1551
Eymin B, Haugg M, Droin N, Sordet O, Dimanche-Boitrel MT, Solary E . P27Kip1 induces drug resistance by preventing apoptosis upstream of cytochrome c release from mitochondria and procaspase-3 activation Oncogene 1999 18: 1411–1418
Torii S, Egan DA, Evans RA, Reed JC . Human Daxx regulates Fas-induced apoptosis from nuclear PML oncogenic domains (PODs) EMBO J 1999 18: 6037–6049
Hannun YA . Functions of ceramide in coordinating cellular responses to stress Science 1996 274: 1855–1859
Jarvis WD, Grant S, Kolesnick RN . Ceramide and the induction of apoptosis Clin Cancer Res 1996 2: 1–6
Liu G, Kleine L, Hebert RL . Advances in the signal transduction of ceramide and related sphingolipids Crit Rev Clin Lab Sci 1999 36: 511–573
Jaffrezou JP, Levade T, Bettaieb A, Andrieu N, Bezombes C, Maestre N, Vermeersch S, Rousse A, Laurent G . Daunorubicin-induced apoptosis: triggering of ceramide generation through sphingomyelin hydrolysis EMBO J 1996 15: 2417–2424
Bettaieb A, Plo I, Mansat-De Mas V, Quillet-Mary A, Levade T, Laurent G, Jaffrezou JP . Daunorubicin- and mitoxantrone-triggered phosphatidylcholine hydrolysis: implication in drug-induced ceramide generation and apoptosis Mol Pharmacol 1999 55: 118–125
Zhang J, Alter N, Reed JC, Borner C, Obeid LM, Hannun YA . Bcl-2 interrupts the ceramide-mediated pathway of cell death Proc Natl Acad Sci USA 1996 93: 5325–5328
Strum JC, Small GW, Pauig SB, Daniel LW . 1-beta-D-arabinofuranosylcytosine stimulates ceramide and diglyceride formation in HL-60 cells J Biol Chem 1994 269: 15493–15497
Quintans J, Kilkus J, McShan CL, Gottschalk AR, Dawson G . Ceramide mediates the apoptotic response of WEHI 231 cells to anti-immunoglobulin, corticosteroids and irradiation Biochem Biophys Res Commun 1994 202: 710–714
Tepper CG, Jayadev S, Liu B, Bielawska A, Wolff R, Yonehara S, Hannun YA, Seldin MF . Role of ceramide as an endogenous mediator of Fas-induced cytotoxicity Proc Natl Acad Sci USA 1995 92: 8443–8447
Bettaieb A, Record M, Come MG, Bras AC, Chap H, Laurent G, Jaffrezou JP . Opposite effects of tumor necrosis factor alpha on the sphingomyelin-ceramide pathway in two myeloid leukemia cell lines: role of transverse sphingomyelin distribution in the plasma membrane Blood 1996 88: 1465–1472
Verheij M, Bose R, Lin XH, Yao B, Jarvis WD, Grant S, Birrer MJ, Szabo E, Zon LI, Kyriakis JM, Haimovitz-Friedman A, Fuks Z, Kolesnick RN . Requirement for ceramide-initiated SAPK/JNK signalling in stress-induced apoptosis Nature 1996 380: 75–79
Mansat V, Laurent G, Bettaieb A, Levade T, Jaffrézou JP . The protein kinase C activators phorbol esters and phosphatidylserines inhibit neutral sphingomyelinase activation, ceramide and apoptosis triggered by daunorubicin Cancer Res 1997 57: 5300–5304
Grant S, Jarvis WD . Modulation of drug-induced by interruption of the protein kinase C signal transduction pathway: a new therapeutic strategy Clin Cancer Res 1996 2: 1915–1920
Jaffrézou JP, Bettaieb A, Levade T, Laurent G . Antitumor-agent-induced apoptosis in myeloid leukemia cells: a controlled suicide Leuk Lymphoma 1998 29: 453–463
Plo I, Bettaieb A, Payrastre B, Mansat-De Mas V, Bordier C, Rousse A, Kowalski-Chauvel A, Laurent G, Lautier D . The phosphoinositide 3-kinase/Akt pathway is activated by daunorubicin in human acute myeloid leukemia cell lines FEBS Lett 1999 452: 150–154
Takeda H, Matozaki T, Takada T, Noguchi T, Yamao T, Tsuda M, Ochi F, Fukunaga K, Inagaki K, Kasuga M . PI 3-kinase γ and protein kinase ξ mediate RAS-independent activation of MAP kinase by a Gi protein-coupled receptor EMBO J 1999 18: 386–395
Moscat J, Diaz-Meco MT . Zeta protein kinase C: a new target for antiproliferative interactions Anti-cancer drugs 1996 7: 143–148
Creagh EM, Sheehan D, Cotter TG . Heat shock proteins – modulators of apoptosis in tumor cells Leukemia 2000 14: 1161–1173
Azuma T, Koths K, Flanagan L, Kwiatkowski D . Gelsolin in complex with phosphatidylinositol 4,5-bisphosphate inhibits caspase-3 and -9 to retard apoptotic progression J Biol Chem 2000 275: 3761–3766
Eymin B, Sordet O, Droin N, Munsch B, Haugg M, Van de Craen M, Vandenabeele P, Solary E . Caspase-induced proteolysis of the cyclin-dependent kinase inhibitor p27Kip1 mediates its anti-apoptotic activity Oncogene 1999 18: 4839–4847
Acknowledgements
Our group is supported by grants from INSERM, the Ligue Nationale Contre le Cancer (comittees: Côte d'Or, Saône et Loire, Nièvre), the Association pour la Recherche sur le Cancer (No. 9567), the Association pour la Recherche sur la Transfusion (ART) and the Association Régionale pour l'Enseignement et la Recherche Scientifique et Technologique en Champagne-Ardenne (ARERS). ND is the recipient of a grant from the Société Française d'Hématologie.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Solary, E., Droin, N., Bettaieb, A. et al. Positive and negative regulation of apoptotic pathways by cytotoxic agents in hematological malignancies. Leukemia 14, 1833–1849 (2000). https://doi.org/10.1038/sj.leu.2401902
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.leu.2401902
Keywords
This article is cited by
-
Ruthenium(II)-N-alkyl phenothiazine complexes as potential anticancer agents
JBIC Journal of Biological Inorganic Chemistry (2018)
-
Antiproliferative activity and apoptosis-inducing mechanism of constituents from Toona sinensis on human cancer cells
Cancer Cell International (2013)
-
Role of Bcl-2 family proteins and caspases in the regulation of apoptosis
Molecular and Cellular Biochemistry (2011)
-
The relationship between cell apoptosis and dephosphorylated RB protein and proliferating cell nuclear antigen in human breast cancer
The Chinese-German Journal of Clinical Oncology (2010)
-
Experimental study on the mechanism of reversal of leukemia multidrug resistance by proteasome inhibitor bortezomib
Clinical Oncology and Cancer Research (2010)
