
Abstract
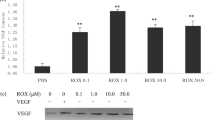
Vascular endothelial growth factor (VEGF) is a potent signalling molecule that acts through two tyrosine kinase receptors, VEGFR1 and VEGFR2. The upregulation of VEGF and its receptors is important in tumour-associated angiogenesis; however, recent studies suggest that several tumour cells express VEGF receptors and may be influenced by autocrine VEGF signalling. Rhabdomyosarcoma (RMS) is the most common paediatric soft-tissue sarcoma and is dependent on autocrine signalling for its growth. The alveolar subtype of RMS is often characterized by the presence of a PAX3-FKHR translocation and when introduced into non-RMS cells, the resultant fusion protein induces expression of VEGFR1. In our study, we examined the expression of VEGF and its receptors in RMS and autocrine effects of VEGF on cell growth. VEGF and receptor mRNA and protein were found to be expressed in RMS cells. Exogenous VEGF addition resulted in extracellular signal-regulated kinase-1/2 phosphorylation and cell proliferation and both were reduced by VEGFR1 blockade. Growth was also slowed by VEGFR1 inhibitor alone. Treatment of RMS cells with all-trans-retinoic acid decreased VEGF secretion and slowed cell growth, which was rescued by VEGF. These data suggest that autocrine VEGF signalling likely influences RMS growth and its inhibition may be an effective treatment for RMS.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others

Abbreviations
- VEGF:
-
vascular endothelial growth factor
- rhVEGF165:
-
recombinant human vascular endothelial growth factor (165 amino-acid isoform)
- PlGF:
-
placenta growth factor
- VEGFR:
-
vascular endothelial growth factor receptor
- flt1:
-
fms-like tyrosine kinase receptor (or VEGFR1)
- KDR:
-
kinase insert domain-containing receptor (or VEGFR2)
- NRP:
-
neuropilin
- RMS:
-
rhabdomyosarcoma
- ARMS:
-
alveolar rhabdomyosarcoma
- ERMS:
-
embryonal rhabdomyosarcoma
- AtRA:
-
all-trans-retinoic acid
- IGF-II:
-
insulin-like growth factor-II
- bFGF:
-
basic fibroblast growth factor
- EGF:
-
epidermal growth factor
- TGF-β:
-
transforming growth factor
- ERK:
-
extracellular signal-regulated kinase
References
Barber TD, Barber MC, Tomescu O, Barr FG, Ruben S and Friedman TB . (2002). Genomics, 79, 278–284.
Barr FG, Fitzgerald JC, Ginsberg JP, Vanella ML, Davis RJ and Bennicelli JL . (1999). Cancer Res., 59, 5443–5448.
Barr FG, Galili N, Holick J, Biegel JA, Rovera G and Emanuel BS . (1993). Nat. Genet., 3, 113–117.
Bates RC, Goldsmith JD, Bachelder RE, Brown C, Shibuya M, Oettgen P and Mercurio AM . (2003). Curr. Biol., 13, 1721–1727.
Beierle EA, Dai W, Langham Jr MR, Copeland III EM and Chen MK . (2003). J. Pediatr. Surg., 38, 514–521.
Beierle EA, Dai W, Langham Jr MR, Copeland III EM and Chen MK . (2004). J. Surg. Res., 119, 56–65.
Borgstrom P, Hillan KJ, Sriramarao P and Ferrara N . (1996). Cancer Res., 56, 4032–4039.
Brodowicz T, Wiltschke C, Kandioler-Eckersberger D, Grunt TW, Rudas M, Schneider SM, Hejna M, Budinsky A and Zielinski CC . (1999). Br. J. Cancer, 80, 1350–1358.
Broggini M, Marchini SV, Galliera E, Borsotti P, Taraboletti G, Erba E, Sironi M, Jimeno J, Faircloth GT, Giavazzi R and D’Incalci M . (2003). Leukemia, 17, 52–59.
Brower V . (2003). J. Natl. Cancer Inst., 95, 1425–1427.
Claesson-Welsh L . (2003). Biochem. Soc. Trans., 31, 20–24.
Crouch GD and Helman LJ . (1991). Cancer Res., 51, 4882–4887.
Damert A, Miquerol L, Gertsenstein M, Risau W and Nagy A . (2002). Development, 129, 1881–1892.
Das B, Yeger H, Tsuchida R, Torkin R, Gee MFW, Thorner P, Ho M, Shibuya M, Malkin D and Baruchel S . Cancer Res. (in press).
Davis RJ, D’Cruz CM, Lovell MA, Biegel JA and Barr FG . (1994). Cancer Res., 54, 2869–2872.
De Giovanni C, Melani C, Nanni P, Landuzzi L, Nicoletti G, Frabetti F, Griffoni C, Colombo MP and Lollini PL . (1995). Br. J. Cancer, 72, 1224–1229.
de Vries C, Escobedo JA, Ueno H, Houck K, Ferrara N and Williams LT . (1992). Science, 255, 989–991.
del Rincon SV, Rousseau C, Samanta R and Miller Jr WH . (2003). Oncogene, 22, 3353–3360.
Dias S, Hattori K, Heissig B, Zhu Z, Wu Y, Witte L, Hicklin DJ, Tateno M, Bohlen P, Moore MA and Rafii S . (2001). Proc. Natl. Acad. Sci. USA, 98, 10857–10862.
Dias S, Hattori K, Zhu Z, Heissig B, Choy M, Lane W, Wu Y, Chadburn A, Hyjek E, Gill M, Hicklin DJ, Witte L, Moore MA and Rafii S . (2000). J. Clin. Invest., 106, 511–521.
Dias S, Shmelkov SV, Lam G and Rafii S . (2002). Blood, 99, 2532–2540.
Diaz BV, Lenoir MC, Ladoux A, Frelin C, Demarchez M and Michel S . (2000). J. Biol. Chem., 275, 642–650.
El-Badry OM, Minniti C, Kohn EC, Houghton PJ, Daughaday WH and Helman LJ . (1990). Cell Growth Differ., 1, 325–331.
Fan F, Wey JS, McCarty MF, Belcheva A, Liu W, Bauer TW, Somcio RJ, Wu Y, Hooper A, Hicklin DJ and Ellis LM . (2005). Oncogene, 24, 2647–2653.
Ferrara N . (1996). Eur. J. Cancer, 32A, 2413–2422.
Folkman J . (1995). Nat. Med., 1, 27–31.
Fong GH, Rossant J, Gertsenstein M and Breitman ML . (1995). Nature, 376, 66–70.
Fuh G, Garcia KC and de Vos AM . (2000). J. Biol. Chem., 275, 26690–26695.
Gille H, Kowalski J, Yu L, Chen H, Pisabarro MT, Davis-Smyth T and Ferrara N . (2000). EMBO J., 19, 4064–4073.
Holash J, Davis S, Papadopoulos N, Croll SD, Ho L, Russell M, Boland P, Leidich R, Hylton D, Burova E, Ioffe E, Huang T, Radziejewski C, Bailey K, Fandl JP, Daly T, Wiegand SJ, Yancopoulos GD and Rudge JS . (2002). Proc. Natl. Acad. Sci. USA, 99, 11393–11398.
Isner JM and Losordo DW . (1999). Nat. Med., 5, 491–492.
Itokawa T, Nokihara H, Nishioka Y, Sone S, Iwamoto Y, Yamada Y, Cherrington J, McMahon G, Shibuya M, Kuwano M and Ono M . (2002). Mol. Cancer Ther., 1, 295–302.
Joukov V, Pajusola K, Kaipainen A, Chilov D, Lahtinen I, Kukk E, Saksela O, Kalkkinen N and Alitalo K . (1996). EMBO J., 15, 290–298.
Kabbinavar F, Hurwitz HI, Fehrenbacher L, Meropol NJ, Novotny WF, Lieberman G, Griffing S and Bergsland E . (2003). J. Clin. Oncol., 21, 60–65.
Kalebic T, Tsokos M and Helman LJ . (1994). Cancer Res., 54, 5531–5534.
Karkkainen MJ, Saaristo A, Jussila L, Karila KA, Lawrence EC, Pajusola K, Bueler H, Eichmann A, Kauppinen R, Kettunen MI, Yla-Herttuala S, Finegold DN, Ferrell RE and Alitalo K . (2001). Proc. Natl. Acad. Sci. USA, 98, 12677–12682.
Ke LD, Fueyo J, Chen X, Steck PA, Shi YX, Im SA and Yung WK . (1998). Int. J. Oncol., 12, 1391–1396.
Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J and Connolly DT . (1989). Science, 246, 1309–1312.
Kendall RL and Thomas KA . (1993). Proc. Natl. Acad. Sci. USA, 90, 10705–10709.
Kendall RL, Wang G and Thomas KA . (1996). Biochem. Biophys. Res. Commun., 226, 324–328.
Kim KJ, Li B, Winer J, Armanini M, Gillett N, Phillips HS and Ferrara N . (1993). Nature, 362, 841–844.
Kini AR, Peterson LA, Tallman MS and Lingen MW . (2001). Blood, 97, 3919–3924.
Kita Y, Tseng J, Horan T, Wen J, Philo J, Chang D, Ratzkin B, Pacifici R, Brankow D, Hu S, Luo Y, Wen D, Arakawa T and Nicolson M . (1996). Biochem. Biophys. Res. Commun., 226, 59–69.
Koufos A, Hansen MF, Copeland NG, Jenkins NA, Lampkin BC and Cavenee WK . (1985). Nature, 316, 330–334.
Langer I, Vertongen P, Perret J, Fontaine J, Atassi G and Robberecht P . (2000). Med. Pediatr. Oncol., 34, 386–393.
Lu D, Jimenez X, Zhang H, Wu Y, Bohlen P, Witte L and Zhu Z . (2001). Cancer Res., 61, 7002–7008.
Luttun A, Tjwa M, Moons L, Wu Y, Angelillo-Scherrer A, Liao F, Nagy JA, Hooper A, Priller J, De Klerck B, Compernolle V, Daci E, Bohlen P, Dewerchin M, Herbert JM, Fava R, Matthys P, Carmeliet G, Collen D, Dvorak HF, Hicklin DJ and Carmeliet P . (2002). Nat. Med., 8, 831–840.
Lyden D, Hattori K, Dias S, Costa C, Blaikie P, Butros L, Chadburn A, Heissig B, Marks W, Witte L, Wu Y, Hicklin D, Zhu Z, Hackett NR, Crystal RG, Moore MA, Hajjar KA, Manova K, Benezra R and Rafii S . (2001). Nat. Med., 7, 1194–1201.
Manley PW, Bold G, Bruggen J, Fendrich G, Furet P, Mestan J, Schnell C, Stolz B, Meyer T, Meyhack B, Stark W, Strauss A and Wood J . (2004). Biochim. Biophys. Acta, 1697, 17–27.
Mercurio AM, Bachelder RE, Bates RC and Chung J . (2004). Semin. Cancer Biol., 14, 115–122.
Merlino G and Helman LJ . (1999). Oncogene, 18, 5340–5348.
Millauer B, Longhi MP, Plate KH, Shawver LK, Risau W, Ullrich A and Strawn LM . (1996). Cancer Res., 56, 1615–1620.
Narendran A, Ganjavi H, Morson N, Connor A, Barlow JW, Keystone E, Malkin D and Freedman MH . (2003). Exp. Hematol., 31, 693–701.
Neufeld G, Cohen T, Gengrinovitch S and Poltorak Z . (1999). FASEB J., 13, 9–22.
Neufeld G, Kessler O and Herzog Y . (2002). Adv. Exp. Med. Biol., 515, 81–90.
Orlandini M, Marconcini L, Ferruzzi R and Oliviero S . (1996). Proc. Natl. Acad. Sci. USA, 93, 11675–11680.
Parry TJ, Cushman C, Gallegos AM, Agrawal AB, Richardson M, Andrews LE, Maloney L, Mokler VR, Wincott FE and Pavco PA . (1999). Nucleic Acids Res., 27, 2569–2577.
Podar K, Catley LP, Tai YT, Shringarpure R, Carvalho P, Hayashi T, Burger R, Schlossman RL, Richardson PG, Pandite LN, Kumar R, Hideshima T, Chauhan D and Anderson KC . (2004). Blood, 103, 3474–3479.
Podar K, Tai YT, Davies FE, Lentzsch S, Sattler M, Hideshima T, Lin BK, Gupta D, Shima Y, Chauhan D, Mitsiades C, Raje N, Richardson P and Anderson KC . (2001). Blood, 98, 428–435.
Quinn TP, Peters KG, De Vries C, Ferrara N and Williams LT . (1993). Proc. Natl. Acad. Sci. USA, 90, 7533–7537.
Ricaud S, Vernus B, Duclos M, Bernardi H, Ritvos O, Carnac G and Bonnieu A . (2003). Oncogene, 22, 8221–8232.
Risau W . (1997). Nature, 386, 671–674.
Schweigerer L, Neufeld G, Mergia A, Abraham JA, Fiddes JC and Gospodarowicz D . (1987). Proc. Natl. Acad. Sci. USA, 84, 842–846.
Selvaraj SK, Giri RK, Perelman N, Johnson C, Malik P and Kalra VK . (2003). Blood, 102, 1515–1524.
Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML and Schuh AC . (1995). Nature, 376, 62–66.
Suzuki K, Hayashi N, Miyamoto Y, Yamamoto M, Ohkawa K, Ito Y, Sasaki Y, Yamaguchi Y, Nakase H, Noda K, Enomoto N, Arai K, Yamada Y, Yoshihara H, Tujimura T, Kawano K, Yoshikawa K and Kamada T . (1996). Cancer Res., 56, 3004–3009.
Tammela T, Enholm B, Alitalo K and Paavonen K . (2005). Cardiovasc. Res., 65, 550–563.
Wakiya K, Begue A, Stehelin D and Shibuya M . (1996). J. Biol. Chem., 271, 30823–30828.
Waltenberger J, Claesson-Welsh L, Siegbahn A, Shibuya M and Heldin CH . (1994). J. Biol. Chem., 269, 26988–26995.
Weninger W, Rendl M, Mildner M and Tschachler E . (1998). J. Invest. Dermatol., 111, 907–911.
Wey JS, Stoeltzing O and Ellis LM . (2004). Clin. Adv. Hematol. Oncol., 2, 37–45.
Willett CG, Boucher Y, di Tomaso E, Duda DG, Munn LL, Tong RT, Chung DC, Sahani DV, Kalva SP, Kozin SV, Mino M, Cohen KS, Scadden DT, Hartford AC, Fischman AJ, Clark JW, Ryan DP, Zhu AX, Blaszkowsky LS, Chen HX, Shellito PC, Lauwers GY and Jain RK . (2004). Nat. Med., 10, 145–147.
Young Jr JL, Ries LG, Silverberg E, Horm JW and Miller RW . (1986). Cancer, 58, 598–602.
Zachary I and Gliki G . (2001). Cardiovasc. Res., 49, 568–581.
Zachary I, Mathur A, Yla-Herttuala S and Martin J . (2000). Arterioscler. Thromb. Vasc. Biol., 20, 1512–1520.
Zhang L, Zhan S, Navid F, Li Q, Choi YH, Kim M, Seth P and Helman LJ . (1998). Oncogene, 17, 1261–1270.
Ziegler BL, Valtieri M, Porada GA, De Maria R, Muller R, Masella B, Gabbianelli M, Casella I, Pelosi E, Bock T, Zanjani ED and Peschle C . (1999). Science, 285, 1553–1558.
Acknowledgements
This work was funded by the Andrew Mizzoni Cancer Research Fund and the James Birrell Research Fund and through studentships of the Ontario Student Opportunity Trust Fund – Hospital for Sick Children Foundation Student Scholarship Program (MG, BD) and the Brainchild Foundation (BD). RT is supported in part by the Scotiabank Clinician-Scientist Training Program Fund and the Research Training Centre, Hospital for Sick Children, Toronto. DM is a Research Scientist of the National Cancer Institute of Canada/Canadian Cancer Society. We are grateful for helpful discussions with Drs Herman Yeger and Meredith Irwin.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Gee, M., Tsuchida, R., Eichler-Jonsson, C. et al. Vascular endothelial growth factor acts in an autocrine manner in rhabdomyosarcoma cell lines and can be inhibited with all-trans-retinoic acid. Oncogene 24, 8025–8037 (2005). https://doi.org/10.1038/sj.onc.1208939
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1208939
Keywords
This article is cited by
-
Evaluation of Endoglin (CD105) expression in pediatric rhabdomyosarcoma
BMC Cancer (2018)
-
Clinicopathological characteristics and treatment outcomes of Chinese patients with genitourinary embryonal rhabdomyosarcoma
World Journal of Surgical Oncology (2015)
-
Proof-of-concept rare cancers in drug development: the case for rhabdomyosarcoma
Oncogene (2014)
-
BMP4/Thrombospondin-1 loop paracrinically inhibits tumor angiogenesis and suppresses the growth of solid tumors
Oncogene (2014)
-
Targeted Therapy in Bone and Soft Tissue Sarcoma in Children and Adolescents
Current Oncology Reports (2012)
