
Abstract
We previosuly described the oral-esophageal tissue-specific expression of cyclin D1 with the Epstein – Barr virus ED-L2 promoter in transgenic mice, and resulting dysplasia. Given the evidence for an interplay between environmental and genetic factors in esophageal squamous carcinogenesis, the aim of this study was to determine the potential cooperation of the nitrosamine compound N-nitrosomethylbenzylamine (NMBA), an esophageal specific carcinogen, in the cyclin D1 transgenic mice. NMBA was first demonstrated to induce dysplasia in two strains of inbred mice, C57BL/6 and FVB/N. Subcutaneous NMBA was then administrated to wild type and transgenic mice beginning at 4 weeks of age. Mice were monitered for the duration of the study for general appearance, activity and weight, and were euthanized at 12 and 15 months. Histopathologic analysis revealed increased severity of dysplasia in cyclin D1 mice treated with NMBA compared with treated age-matched wild-type mice and untreated mice. There was also increased proliferating cell nuclear antigen (PCNA) expression in the esophagi of NMBA treated cyclin D1 mice. Taken together, these findings suggest that a genetic alteration, specifically cyclin D1 overexpression and a chemical carinogen, NMBA, may cooperate to increase the severity of esophageal squamous dysplasia, a prominent precursor to carcinoma.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
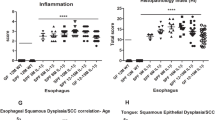

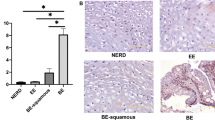
References
Albert JM, Munger K, Howley PM and Hanahan D. . 1994 J. Virol. 68,: 4358–4368.
Antrup H and Stoner GD. . 1982 Cancer Res. 42: 1307–1311.
Balmain A and Brown K. . 1988 Adv. Cancer Res. 51: 147–182.
Barch DH, Walloch H, Hedvegi D and Iannaccone PM. . 1986 J. Natl. Cancer. Inst. 77: 1145–1153.
Bianchi AB, Fischer SM, Robles AI, Rinchik EM and Conti CJ. . 1993 Oncogene 8: 1127–1133.
Brown K, Buchmann A and Balmain A. . 1990 Proc. Natl. Acad. Sci. USA 87: 538–542.
Craddock VM and Driver HE. . 1987 Carcinogenesis 8: 1129–1132.
Donehower LA, Harvey M, Siagle B, McArthur MJ, Montgomery Jr CA, Butel JS and Bradley A. . 1992 Nature 356: 215–221.
Doniger J, Day RS and Dipaolo JA. . 1985 Proc. Natl. Acad. Sci. USA 82: 421–425.
Druckrey H, Preussmann R, Blum G, Ivankovic S and Afkham J. . 1963 Naturwissenschaften 50: 100–101.
Fong LYY, Lin HT and Lee CLH. . 1979 Int. J. Cancer 23: 679–682.
Garber SA, Fernstrom MJ, Stoner GD and Ruch RJ. . (1997) Carcinogenesis 18: 1149–1153.
Harris CC, Antrup H, Stoner GD, Trump BF, Hillman E, Schafer PW and Jeffrey AM. . 1979 Cancer Res. 39: 4401–4406.
Hodgson RM, Schweinsberg F, Wiessler M and Kleihues P. . 1982 Cancer Res. 42: 2836–2840.
Jiang W, Kahn SM, Tomita N, Zhang YJ, Lu SH, Weinstein IB. . 1992 Cancer Res. 52: 2980–2983.
Jiang W, Zhang YU, Kahn S, Hollstein MC, Santella RM, Lu SH, Harris CC, Montesano R and Weinstein IB. . 1993 Proc. Natl. Acad. Sci. USA 90: 9026–9030.
Kemp CJ, Donehower LA, Bradley A and Balmain A. . 1993 Cell 74: 813–822.
Labuc GE and Archer MC. . 1982 Cancer Res. 42: 3181–3186.
Li MH, Ji C and Cheng SJ. . 1986 Nutr. Cancer 8: 63–69.
Lijinsky W, Saavedra JE, Rueber MD and Singer SS. . 1982 J. Natl. Cancer. Inst. 68: 681–684.
Mitsunaga SI, Zhang SY, Ruggeri BA, Giminez-Conti I, Robles AI, Conti CJ and Klein-Szanto AJP. . 1995 Carcinogenesis 16: 1629–1635.
Morse MA and Stoner GD. . 1993 Carcinogenesis 14: 1737–1746.
Mueller A, Odze R, Jenkins TD, Shahsafaei A, Nakagawa H, Inomoto T and Rustgi AK. . 1997 Cancer Res. 57: 5542–5549.
Nakagawa H, Zukerberg L, Togawa K, Meltzer SJ, Nishihara T and Rustgi AK. . 1995 Cancer 76: 541–549.
Nakagawa H, Wang TC, Odze R, Zukerberg LR, Togawa K, May GHW, Wilson J and Rustgi AK. . 1997 Oncogene 14: 1185–1190.
Odeleye OE, Eskelson CD, Mufti SI and Watson RR. . 1992a Ann. NY Acad. Sci. 669: 368–370.
Odeleye OE, Eskelson CD, Mufti SI and Watson RR. . 1992b Carcinogenesis 13: 1811–1816.
Quintanilla M, Brown K, Ramsden M and Balmain A. . 1986 Nature 322: 78–80.
Robles AI and Conti CJ. . 1995 Carcinogenesis 16: 781–786.
Sheyn I, Noffsinger AE, Heffelfinger S, Davis B, Miller MA and Fenoglio-Preiser CM. . 1997 Human Path. 28: 270–276.
Siglin JC, Morse MA, Schut HAJ, Geil RG, Conran PB and Stoner GD. . 1996 Carcinogenesis 17: 1135–1140.
Stern MC, Giminez-Conti IB and Conti CJ. . 1995 Carcinogenesis 16: 1947–1953.
Stinson SF, Squire RA and Sporn MB. . 1978 J. Natl. Cancer Inst. 61: 1471–1473.
Van Benthem J, Vermeulen E, Winterwerp HHK, Wild CP, Scherer E and Den Engelse L. . 1992 Carcinogenesis 11: 2101–2105.
Walker EA, Castegnaro M, Garren L, Toussaint G and Kowalski B. . 1979 J. Natl. Cancer Inst. 63: 947–951.
Wang D, Weghorst CM, Calvert RJ and Stoner GD. . 1996 Carcinogenesis 17: 625–630.
Wang OS, Sabourin CL, Bijur GN, Robertson FH and Stoner GD. . 1996a Mol. Carcinog. 15: 144–153.
Wang OS, Sabourin CLK, Wang H and Stoner GD. . 1996b Carcinogenesis 17: 1583–1588.
Wang Y, You M, Reynolds SH, Stoner GD and Anderson MW. . 1990 Cancer Res. 50: 1591–1595.
Yan YX, Nakagawa H, Lee MH, and Rustgi AK. . 1997 J. Biol. Chem. 52: 33181–33190.
Youssef EM, Hasuma T, Morishima Y, Takada N, Osugi H, Higashino M, Otani S and Fukushima S. . 1997 Japan. J. Cancer Res. 88: 18–25.
Acknowledgements
This work was supported by the ADHF/AGA Funderberg Awards (AR), an ACS Jr Faculty Research Award (AR), NIH DK53377 (AR), NIH P01 DE12467-01A1 (AR, RK), a Glaxo Wellcome Institute for Digestive Health Basic Research Award (TJ), an NIDDK K08 Award (TJ) and a Center for the Study of Inflammatory Bowel Disease Award from NIH 5P30DK 43357-08 (TJ). A. Mueller was supported by the Deutsche Forschungsgemeinschaft (DFG #1304-1-1). R. Odze was supported by the Stanley L. Robbins Research Fund from the Department of Pathology at Brigham and Women's Hospital.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Jenkins, T., Mueller, A., Odze, R. et al. Cyclin D1 overexpression combined with N-nitrosomethylbenzylamine increases dysplasia and cellular proliferation in murine esophageal squamous epithelium. Oncogene 18, 59–66 (1999). https://doi.org/10.1038/sj.onc.1202296
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1202296
Keywords
This article is cited by
-
Increased susceptibility to the tobacco carcinogen 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone in transgenic mice overexpressing c-myc and epidermal growth factor in alveolar type II cells
Journal of Cancer Research and Clinical Oncology (2003)
-
Ectopic expression of cyclin D1 impairs the proliferation and enhances the apoptosis of a murine lymphoid cell line
Cell Death & Differentiation (2001)
