
Abstract
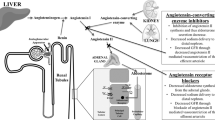
Although the angiotensin receptor antagonist (ARB) shares the angiotensin-II-blocking activity with the angiotensin-converting enzyme inhibitor (ACE-I), pharmacological mechanisms of action of these agents differ. We evaluated the temporal profiles of action of ACE-I and ARB on urinary protein excretion and nitrate/nitrate (NOx) excretion in hypertensive (140 and/or 90 mmHg) patients with chronic renal disease (serum creatinine<265 (range, 44–265) μmol/l or creatinine clearance>30 (range, 30–121) ml/min). Patients with mild (<1 g/day; range, 0.4–1.0) and moderate proteinuria (>1 g/day; range, 1.1–6.9) were randomly assigned to ACE-I- and ARB-treated groups, and were treated with ACE-I (trandolapril or perindopril) or ARB(losartan or candesartan) for 48 weeks. In all groups, treatment with ACE-I or ARB decreased blood pressure to the same level, but had no effect on creatinine clearance. In patients with mild proteinuria, neither ACE-I nor ARB altered urinary protein excretion. In patients with moderate proteinuria, ACE-I caused 44±6% reduction in proteinuria (from 2.7±0.5 to 1.5±0.4 g/day, n=14) at 12 weeks, and this beneficial effect persisted throughout the protocol (48 weeks, 1.2±0.2 g/day). In contrast, ARB did not produce a significant decrease in proteinuria at 12 weeks (23±8%, n=13), but a 41±6% reduction in proteinuria was observed at 48 weeks. Similarly, although early (12 weeks) increases in urinary NOx excretion were observed with ACE-I (from 257±70 to 1111±160 μmol/day) and ARB (from 280±82 to 723±86 μmol/day), the ARB-induced increase in NOx excretion was smaller than that by ACE-I (P<0.05). In conclusion, although both ACE-I and ARB reduce blood pressure similarly, the effect of these agents on proteinuria differs in chronic renal disease with moderate proteinuria. Relatively early onset of the proteinuria-reducing effect was observed with ACE-I, which paralleled the increase in urinary NOx excretion. Conversely, ARB decreased proteinuria and increased urinary NOx excretion gradually. These time course-dependent changes in proteinuria and urinary NOx may reflect the pharmacological property of ACE-I and ARB, with regard to the action on bradykinin.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 digital issues and online access to articles
$119.00 per year
only $9.92 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
References
Anderson S, Rennke HG, Brenner BM . Therapeutic advantage of converting enzyme inhibitors in arresting progressive renal disease associated with systemic hypertension in the rat. J Chin Invest 1986; 77: 1993–2000.
Taguma Y et al. Effect of captopril on heavy proteinuria in azotemic diabetics. N Engl J Med 1985; 313: 1617–1620.
Lewis EJ, Hunsicker LG, Bain RP, Rohde RD . The effect of angiotensin-converting enzyme inhibition on diabetic nephropathy. N Engl J Med 1993; 326: 1456–1462.
Maschio G et al. AIPRI Study Group. effect of the angiotensin-converting enzyme inhibitor benazepril on the progression of chronic renal insufficiency. N Engl J Med 1996; 334: 939–945.
Brenner BM et al. Effects of losartan on renal and cardiovascular outcomes in patients with type 2 diabetes and nephropathy. N Engl J Med 2001; 345: 861–869.
Lewis EJ et al. Collaborative Study Group. Renoprotective effect of the angiotensin-receptor antagonist irbesartan in patients with nephropathy due to type 2 diabetes. N Engl J Med 2001; 345: 851–860.
Kon V, Fogo A, Ichikawa I . Bradykinin causes selective efferent arteriolar dilation during angiotensin I converting enzyme inhibition. Kidney Int 1993; 44: 545–550.
Matsuda H et al. Zonal heterogeneity in action of angiotensin-converting enzyme inhibitor on renal microcirculation: role of intrarenal bradykinin. J Am Soc Nephrol 1999; 10: 2272–2282.
Gainer JV et al. Effect of bradykinin-receptor blockade on the response to angiotensin-converting-enzyme inhibitor in normotensive and hypertensive subjects. N Engl J Med 1998; 339: 1285–1292.
Siragy HM, Jaffa AA, Margolius HS, Carey RM: Renin–angiotensin system modulates renal bradykinin production. Am J Physiol 1996; 271: R1090–R1095.
Nishimoto M et al. Significance of chymase-dependent angiotensin II-forming pathway in the development of vascular proliferation. Circulation 2001; 104: 1274–1279.
Russo D et al. Coadministration of losartan and enalapril exerts additive antiproteinuric effect in IgA nephropathy. Am J Kidney Dis 2001; 38: 18–25.
Tokuyama H et al. Role of intrarenal angiotensin II (AII) and cyclooxygenase (COX)-1/2 in chronic ischemic nephropathy. J Hypertens 2000; 18 (Suppl 4): S12.
Ohta K et al. A novel in vivo assay system for consecutive measurement of brain nitric oxide production combined with the microdialysis technique. Neurosci Lett 1994; 176: 165–168.
Liu Y-H et al. Effects of angiotensin-converting enzyme inhibitors and angiotensin II type 1 receptor antagonists in rats with heart failure. Role of kinins and angiotensin II type 2 receptors. J Clin Invest 1997; 99: 1926–1935.
Carey RM, Jin X, Wang Z, Siragy HM . Nitric oxide: a physiological mediator of the type 2 (AT2) angiotensin receptor. Acta Physiol Scand 2000; 168: 65–71.
Adler L et al. Angiotensin converting enzyme inhibitor therapy in children with Alport syndrome: effect on urinary albumin, TGF-B, and nitrite excretion. BMC Nephrol 2002; 3: 2.
Rahma M et al. Effects of furosemide on the tubular reabsorption of nitrates in anesthetized dogs. Eur J Pharmacol 2001; 428: 113–119.
Sosa-Canache B, Cierco M, Gutierrez CI, Israel A . Role of bradykinin and nitric oxide in the AT2 receptor-mediated hypotension. J Hum Hypertens 2000; 14: 4S40–4S46.
Siragy HM, de Gasparo M, Carey RM . Angiotensin type 2 receptor mediates valsartan-induced hypotension in conscious rats. Hypertension 2000; 35: 1074–1077.
Weir MR, Henrich WL . Theoretical basis and clinical evidence for differential effects of angiotensin-converting enzyme inhibitors and angiotensin II receptor subtype 1 blockers. Curr Opin Nephrol Hypertens 2000; 9: 403–411.
Ehara T, Shigematsu H . Contribution of mast cells to the tubulointerstitial lesions in IgA nephritis. Kidney Int 1998; 54: 1675–1683.
Ghiadoni L et al. Effect of the angiotensin II type 1 receptor blocker candesartan on endothelial function in patients with essential hypertension. Hypertension 2000; 35 (1 Part 2): 501–506.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Matsuda, H., Hayashi, K. & Saruta, T. Distinct time courses of renal protective action of angiotensin receptor antagonists and ACE inhibitors in chronic renal disease. J Hum Hypertens 17, 271–276 (2003). https://doi.org/10.1038/sj.jhh.1001543
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.jhh.1001543
Keywords
This article is cited by
-
Updated Report on Comparative Effectiveness of ACE inhibitors, ARBs, and Direct Renin Inhibitors for Patients with Essential Hypertension: Much More Data, Little New Information
Journal of General Internal Medicine (2012)
-
Beneficial action of candesartan cilexetil plus amlodipine or ACE inhibitors in chronic nondiabetic renal disease
Journal of Human Hypertension (2004)
