
Abstract
The analyses of large epidemiological databases have suggested that infants and children who show catch-up growth, or adiposity rebound at a younger age, are predisposed to the development of obesity, type 2 diabetes and cardiovascular diseases later in life. The pathophysiological mechanisms by which these growth trajectories confer increased risks for these diseases are obscure, but there is compelling evidence that the dynamic process of catch-up growth per se, which often overlaps with adiposity rebound at a younger age, is characterized by hyperinsulinemia and by a disproportionately higher rate in the recovery of body fat than lean tissue (i.e. preferential ‘catch-up fat’). This paper first focuses upon the almost ubiquitous nature of this preferential ‘catch-up fat’ phenotype across the life cycle as a risk factor for obesity and insulin-related complications – not only in infants and children who experienced catch-up growth after earlier fetal or neonatal growth retardation, or after preterm birth, but also in adults who show weight recovery after substantial weight loss owing to famine, disease-cachexia or periodic dieting. It subsequently reviews the evidence indicating that such preferential catch-up fat is primarily driven by energy conservation (thrifty) mechanisms operating via suppressed thermogenesis, with glucose thus spared from oxidation in skeletal muscle being directed towards de novo lipogenesis and storage in white adipose tissue. A molecular–physiological framework is presented which integrates emerging insights into the mechanisms by which this thrifty ‘catch-up fat’ phenotype crosslinks with early development of insulin and leptin resistance. In the complex interactions between genetic constitution of the individual, programming earlier in life, and a subsequent lifestyle of energy dense foods and low physical activity, this thrifty ‘catch-up fat’ phenotype – which probably evolved to increase survival capacity in a hunter–gatherer lifestyle of periodic food shortages – is a central event in growth trajectories to obesity and to diseases that cluster into the insulin resistance (metabolic) syndrome.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$259.00 per year
only $21.58 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout






Similar content being viewed by others

References
Hamm P, Shekelle RB, Stamler J . Large fluctuations in body weight during young adulthood and twenty-five-year risk of coronary death in men. Am J Epidemiol 1989; 129: 312–318.
Lissner L, Bengtsson C, Lapidus L, Larsson B, Bengtsson B, Brownell K . Body weight variability and mortality in the Gothenburg prospective studies of men and women. In: Bjorntorp P, Rossner S (eds). Obesity in Europe 88. John Libbey: UK, 1989, pp 55–60.
Holbrook TL, Barrett-Connor E, Wingard D . The association of lifetime weight and weight control patterns with diabetes among men and women in an adult community. Int J Obes Rel Metab Disord 1989; 13: 723–729.
Lissner L, Odell PM, D'Agostino RB, Stokes J, Kreger BE, Belanger AJ et al. Variability of body weight and health outcomes in the Framingham population. N Engl J Med 1991; 324: 1839–1844.
Lee IM, Paffenbarger RS . A 27-year follow-up of middle-aged men. JAMA 1992; 268: 2045–2049.
Diaz VA, Mainous AG, Everett CJ . The association between weight fluctuation and mortality: results from a population-based cohort study. J Commun Health 2005; 30: 153–165.
Montani JP, Viecelli AK, Prévot A, Dulloo AG . Weight cycling during growth and beyond: a risk factor for later cardiovascular diseases. Int J Obes Rel Metab Disord 2006; 30: S58–S66.
Yatsuya H, Tamakoshi K, Yoshida T, Hori Y, Zhang H, Ishikawa M et al. Association between weight fluctuation and fasting insulin concentration in Japanese men. Int J Obes Relat Metab Disord 2003; 27: 478–483.
Zhang H, Tamakoshi K, Yatsuya H, Murata C, Wada K, Otsuka R et al. Long-term body weight fluctuation is associated with metabolic syndrome independent of current body mass index among Japanese men. Circ J 2005; 69: 13–18.
Taeko K, Shigeki T, Hiroshi S, Yuzo S . Effects of weight cycling on young women. Obesity Rev 2006; 7 (Suppl 2): 181 (Abstract).
Eriksson JG, Forsen T, Tuomilehto J, Winter PD, Osmond C, Barker DJ . Catch-up growth in childhood and death from coronary heart disease: longitudinal study. BMJ 1999; 318: 427–431.
Cianfarani S, Germani D, Branca F . Low birth weight and adult insulin resistance: the ‘catch-up growth’ hypothesis. Arch Dis Child Fetal Neonatal Ed 1999; 81: F71–F73.
Huxley RR, Shiell AW, Law CM . The role of size at birth and postnatal catch-up growth in determining systolic blood pressure: a systematic review of the literature. J Hypertens 2000; 18: 815–831.
Ong KK, Ahmed ML, Emmett PM, Preece MA, Dunger DB . Association between postnatal catch-up growth and obesity in childhood: prospective cohort study. BMJ 2000; 320: 967–971.
Levy-Marchal C, Jaquet D, Czernichow P . Long-term metabolic consequences of being born small for gestational age. Semin Neonatol 2000; 9: 67–74.
Barker DJ, Osmond C, Forsen TJ, Kajantie E, Eriksson JG . Trajectories of growth among children who have coronary events as adults. N Engl J Med 2005; 353: 1802–1809.
Popkin BM, Richards MK, Monteiro CA . Stunting is associated with overweight in children of four nations that are undergoing nutrition transition. J Nutr 1996; 126: 3009–3016.
Levitt NS, Lambert EV, Woods D, Hales CN, Andrew R, Seckl JR . Impaired glucose tolerance and elevated blood pressure in low birth weight, non-obese, young South African adults: early programming of cortisol axis. J Clin Endocrinol Metabol 2000; 85: 4611–4618.
Victora CG, Barros FC . The catch-up dilemma – relevance of Leitch's ‘low-high’ pig to child growth in developing countries. Int J Epidemiol 2001; 30: 217–220.
Sawaya AL, Martins P, Hoffman D, Roberts SB . The link between childhood undernutrition and risk of chronic diseases in adulthood: a case study of Brazil. Nutr Rev 2003; 61: 168–175.
Bavdekar A, Yajnik CS, Fall CH, Bapat S, Pandit AN, Deshpande V et al. Insulin resistance syndrome in 8-year-old Indian children: small at birth, big at 8 years, or both? Diabetes 1999; 48: 2422–2429.
Singhal A, Fewtrell M, Cole TJ, Lucas A . Low nutrient intake and early growth for later insulin resistance in adolescents born preterm. Lancet 2003; 361: 1089–1097.
Hofman PL, Regan F, Jackson WE, Jefferies C, Knight DB, Robinson EM et al. Premature birth and later insulin resistance. N Engl J Med 2004; 351: 2179–2186.
Euser AM, Finken MJ, Keijzer-Veen MG, Hille ET, Wit JM, Dekker FW . Associations between prenatal and infancy weight gain and BMI, fat mass, and fat distribution in young adulthood: a prospective cohort study in males and females born very preterm. Am J Clin Nutr 2005; 81: 480–487.
Finken MJ, Keijzer-Veen MG, Dekker FW, Frolich M, Hille ET, Romijn JA et al. Preterm birth and later insulin resistance: effects of birth weight and postnatal growth in a population based longitudinal study from birth into adult life: Insulin resistance 19 years after preterm birth. Diabetologia 2006; 49: 478–485.
Rolland-Cachera MF, Deheeger M, Bellisle F, Sempe M, Guilloud-Bataille M, Patois E . Adiposity rebound in children: a simple indicator for predicting obesity. Am J Clin Nutr 1984; 39: 129–135.
Taylor RW, Grant AM, Goulding A, Williams SM . Early adiposity rebound: review of papers linking this to subsequent obesity in children and adults. Curr Opin Clin Nutr Metab Care 2005; 8: 607–612.
Eriksson JG, Forsen T, Tuomilehto J, Osmond C, Barker DJ . Early adiposity rebound in childhood and risk of type 2 diabetes in adult life. Diabetologia 2003; 46: 190–194.
Bhargava SK, Sachdev HS, Fall CH, Osmond C, Lakshmy R, Barker DJ et al. Relation of serial changes in childhood body-mass index to impaired glucose tolerance in young adulthood. N Engl J Med 2004; 350: 865–875.
Wadsworth M, Butterworth S, Marmot M, Ecob R, Hardy R . Early growth and type 2 diabetes: evidence from the 1946 British birth cohort. Diabetologia 2005; 48: 2505–2510.
Rolland-Cachera MF, Deheeger M, Akrout M, Bellisle F . Influence of macronutrients on adiposity development: a follow up study of nutrition and growth from 10 months to 8 years of age. Int J Obes Relat Metab Disord 1995; 19: 573–578.
Hales CN, Barker DJ . The thrifty phenotype hypothesis. Br Med Bull 2001; 60: 5–20.
Young JB . Programming of sympathoadrenal function. Trends Endocrinol Metab 2002; 13: 381–385.
Colle E, Schiff D, Andrew G, Bauer CB, Fitzhardinge P . Insulin responses during catch-up growth of infants who were small for gestational age. Pediatrics 1976; 57: 363–371.
Crowther NJ, Cameroun N, Trusler J, Gray IP . Association between poor glucose tolerance and rapid post natal weight gain in seven-year-old children. Diabetologia 1998; 41: 1163–1167.
Ong KK, Dunger DB . Birth weight, infant growth and insulin resistance. Eur J Endocrinol 2004; 151 (Suppl 3): U131–U139.
Soto N, Bazaes RA, Pena V, Salazar T, Avila A, Iniguez G et al. Insulin sensitivity and secretion are related to catch-up growth in small-for-gestational-age infants at age 1 year: results from a prospective cohort. J Clin Endocrinol Metab 2003; 88: 3645–3650.
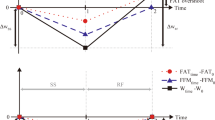
Dulloo AG, Jacquet J, Montani JP . Pathways from weight fluctuations to metabolic diseases: focus on maladaptive thermogenesis during catch-up fat. Int J Obes Relat Metab Disord 2002; 26 (Suppl 2): S46–S57.
Widdowson EM, Shaw WT . Full and empty fat cells. Lancet 1973; 2: 905.
Ashworth A . Growth rates in children recovering from protein-calorie malnutrition. Br J Nutr 1969; 23: 835–845.
Garrow JS, Fletcher K, Halliday D . Body composition in severe infantile malnutrition. J Clin Invest 1965; 44: 417–425.
Graham GG, Cordano A, Blizzard RM, Cheek BD . Infantile malnutrition. Changes in body composition during nutritional rehabilitation. Pediatr Res 1969; 3: 579–589.
MacLean WC, Graham GG . The effect of energy intake on nitrogen content of weight gained by recovering malnourished infant. Am J Clin Nutr 1980; 33: 903–909.
Jackson AA . Nutritional adaptation in disease and recovery. In: Blaxter K, Waterlow JC (eds). Nutritional Adaptation in Man. John Libbey and Company Ltd., London, UK, 1984, pp 111–126.
Castilla-Serna L, Pérez-Ortiz B, Cravioto J . Patterns of muscle and fat mass repair during recovery from advanced infantile protein-energy malnutrition. Eur J Clin Nutr 1996; 50: 392–397.
Benefice E, Garnier D, Simondon KB, Malina RM . Relationship between stunting in infancy and growth and fat distribution during adolescence in Senegalese girls. Eur J Clin Nutr 2001; 55: 50–58.
Martins PA, Hoffman DJ, Fernandes MT, Nascimento CR, Roberts SB, Sesso R et al. Stunted children gain less lean body mass and more fat mass than their non-stunted counterparts: a prospective study. Br J Nutr 2004; 92: 819–825.
Jornayvaz FR, Selz R, Tappy L, Theintz GE . Metabolism of oral glucose in children born small for gestational age: evidence for an impaired whole body glucose oxidation. Metabolism 2004; 53: 847–8851.
Ibanez L, Ong K, Dunger DB, de Zegher F . Early development of adiposity and insulin resistance after catch-up weight gain in small-for-gestational-age children. J Clin Endocrinol Metab 2006; 91: 2153–2158.
Modi N, Thomas EL, Harrington TA, Uthaya S, Dore CJ, Bell JD . Determinants of adiposity during preweaning postnatal growth in appropriately grown and growth-restricted term infants. Pediatr Res 2006; 60: 345–348.
Eriksson J, Forsen T, Tuomilehto J, Osmond C, Barker D . Size at birth, fat-free mass and resting metabolic rate in adult life. Horm Metab Res 2002; 34: 72−76.
Kensara OA, Wootton SA, Phillips DI, Patel M, Jackson AA, Elia M . Fetal programming of body composition: relation between birth weight and body composition measured with dual-energy X-ray absorptiometry and anthropometric methods in older Englishmen. Am J Clin Nutr 2005; 82: 980–987.
Rasmussen EL, Malis C, Jensen CB, Jensen JE, Storgaard H, Poulsen P et al. Altered fat tissue distribution in young adult men who had low birth weight. Diabetes Care 2005; 28: 151–1153.
Hermann TS, Rask-Madsen C, Ihlemann N, Dominguez H, Jensen CB, Storgaard H et al. Normal insulin-stimulated endothelial function and impaired insulin-stimulated muscle glucose uptake in young adults with low birth weight. J Clin Endocrinol Metab 2003; 88: 1252–1257.
Ozanne SE, Jensen CB, Tingey KJ, Storgaard H, Madsbad S, Vaag AA . Low birthweight is associated with specific changes in muscle insulin-signalling protein expression. Diabetologia 2005; 48: 547–552.
Uthaya S, Thomas EL, Hamilton G, Dore CJ, Bell J, Modi N . Altered adiposity after extremely preterm birth. Pediatr Res 2005; 57: 211–215.
Dietz WH . ‘Adiposity rebound’: reality or epiphenomenon? Lancet 2000; 356: 2027–2028.
Williams SM . Weight and height growth rate and the timing of adiposity rebound. Obes Res 2005; 13: 1123–1130.
Taylor RW, Goulding A, Lewis-Barned NJ, Williams SM . Rate of fat gain is faster in girls undergoing early adiposity rebound. Obes Res 2004; 12: 1228–1230.
Jamin F, Müller E . Specific weight of the living man with clinical applications for recovery of body weight. Münchener Medizinische Wochenschrift 1903; 50: 1454–1457; 1511–1515.
Debray C, Zarakovitch M, Ranson B, Jacquemin J, Robert J, Siraga M . Contribution to the study on the pathology of the deportees. Seminaire Hôpital de Paris 1946; 22: 863–870.
Keys A, Brozek J, Henschel A, Mickelson O, Taylor HL . The Biology of Human Starvation. University of Minnesota Press: Minneapolis, MN, 1950.
Dulloo AG, Jacquet J, Girardier L . Poststarvation hyperphagia and body fat overshooting in humans: a role for feedback signals from lean and fat tissues. Am J Clin Nutr 1997; 65: 717–723.
Weyer C, Walford RL, Harper IT, Milner M, MacCallum T, Tataranni PA et al. Energy metabolism after 2 y of energy restriction : the Biosphere 2 experiment. Am J Clin Nutr 2000; 72: 946–953.
Barac-Nieto M, Spurr GB, Lotero H, Maksud MG, Dahners HW . Body composition during nutritional repletion of severely undernourished men. Am J Clin Nutr 1979; 32: 981–991.
Mitchell PB, Truswell AS . Body composition in anorexia nervosa and starvation. In: Beaumont PJV, Burrows GD, Casper RC (eds). Handbook of Eating Disorders. Part I: Anorexia and Bulimia Nervosa. Elsevier: Amsterdam, 1987, pp 45–77.
Van Eys J . Nutrition and cancer. Ann Rev Nutr 1985; 5: 435–461.
Streat SJ, Brodie AH, Hill GL . Aggressive nutritional support does not prevent protein loss despite fat gain in septic intensive care. J Trauma 1987; 27: 262–266.
Kotler DP, Tierney AR, Culpepper-Morgan JA, Wong J, Pierson Jr RM . Effect of parenteral nutrition on body composition in patients with acquired immunodeficiency syndrome. JPEN 1990; 14: 454–458.
Dulloo AG, Montani JP . Obesity in Parkinson's disease patients on electrotherapy: collateral damage, adiposity rebound or secular trends? Br J Nutr 2005; 93: 417–419.
Mayer L, Walsh BT, Pierson Jr RN, Heymsfield SB, Gallagher D, Wang J et al. Body fat redistribution after weight gain in women with anorexia nervosa. Am J Clin Nutr 2005; 81: 1286–1291.
Ounsted M, Sleigh G . The infant's self-regulation of food intake and weight gain. Difference in metabolic balance after growth constraint or acceleration in utero. Lancet 1975; 1 (7922): 1393–1397.
Webster AJF . Energy partitioning, tissue growth and appetite control. Proc Nutr Soc 1993; 52: 69–76.
Dulloo AG, Jacquet J, Girardier L . Autoregulation of body composition during weight recovery in humans : the Minnesota Experiment revisited. Int J Obes 1996; 20: 393–405.
Miller DS, Wise A . The energetics of ‘catch-up’ growth. Nutr Metab 1976; 20: 125–134.
Boyle PC, Storlien LH, Keesey RE . Increased efficiency of food utilization following weight loss. Physiol Behav 1978; 21: 261–264.
Boyle PC, Storlien LH, Harper AE, Keesey RE . Oxygen consumption and locomotor activity during restricted feeding and realimentation. Am J Physiol 1981; 241: R392–R397.
Hill JO, Fried SK, Digirolamo M . Effects of fasting and restricted refeeding on utilization of ingested energy in rats. Am J Physiol 1984; 247: R318–R327.
Dulloo AG, Girardier L . Adaptive changes in energy expenditure during refeeding following low calorie intake: evidence for a specific metabolic component favouring fat storage. Am J Clin Nutr 1990; 52: 415–420.
Dulloo AG, Girardier L . Adaptive role of energy expenditure in modulating body fat and protein deposition during catch-up growth after early undernutrition. Am J clin Nutr 1993; 58: 614–621.
Evans SA, Messina MM, Knight WD, Parsons AD, Overton JM . Long–Evans and Sprague–Dawley rats exhibit divergent responses to refeeding after caloric restriction. Am J Physiol 2005; 288: R1468–R1476.
MacLean PS, Higgins JA, Johnson GC, Fleming-Elder BK, Donahoo WT, Melanson EL et al. Enhanced metabolic efficiency contributes to weight regain after weight loss in obesity-prone rats. Am J Physiol 2004; 287: R1306–R1315.
Dulloo AG, Jacquet J . An adipose-specific control of thermogenesis in body weight regulation. Int J Obes Rel Metab Disord 2001; 25 (Suppl 5): S22–S29.
Dulloo AG, Jacquet J . Adaptive reduction in basal metabolic rate in response to food deprivation in humans: a role for feedback signals from fat stores. Am J Clin Nutr 1998; 68: 599–606.
Foster GD, Wadden TA, Kendrick ZV, Letizia KA, Lander DP, Connill AM . The energy cost of walking before and after significant weight loss. Med Sci Sports Exerc 1995; 27: 888–894.
Kulkarni R, Shetty P . Net mechanical efficiency during stepping in chronically energy deficient human subjects. Ann Hum Biol 1992; 19: 421–425.
Rosenbaum M, Vandenborne K, Goldsmith R, Simoneau JA, Heymsfield S, Joanisse DR et al. Effects of experimental weight perturbation on skeletal muscle work efficiency in human subjects. Am J Physiol 2003; 285: R183–R192.
Ma SW, Foster DO . Starvation-induced changes in metabolic rate, blood flow, and regional energy expenditure in rats. Can J Physiol Pharmacol 1986; 64: 1252–1258.
Dulloo AG . A role for suppressed skeletal muscle thermogenesis in pathways from weight fluctuations to the insulin resistance syndrome. Acta Physiol Scand 2005; 184: 295–307.
Crescenzo R, Samec S, Antic V, Rohner-Jeanrenaud F, Seydoux J, Montani JP et al. A role for suppressed thermogenesis favouring catch-up fat in the pathophysiology of catch-up growth. Diabetes 2003; 52: 1090–1097.
Cettour-Rose P, Samec S, Russell AP, Summermatter S, Mainieri D, Carrillo-Theander C et al. Redistribution of glucose from skeletal muscle to adipose tissue during catch-up fat: A link between catch-up growth and later metabolic syndrome. Diabetes 2005; 54: 751–756.
Dulloo AG, Stock MJ, Solinas G, Boss O, Montani JP, Seydoux J . Leptin directly stimulates thermogenesis in skeletal muscle. FEBS Lett 2002; 515: 109–113.
Solinas G, Summermatter S, Mainieri D, Gubler M, Pirola L, Wymann MP et al. The direct effect of leptin on skeletal muscle thermogenesis is mediated by substrate cycling between de novo lipogenesis and lipid oxidation. FEBS Lett 2004; 577: 539–544.
Solinas G, Summermatter S, Mainieri D, Gubler M, Montani JP, Seydoux J et al. Corticotropin-releasing hormone directly stimulates thermogenesis in skeletal muscle possibly through substrate cycling between de novo lipogenesis and lipid oxidation. Endocrinology 2006; 147: 31–38.
Summermatter S, Mainieri D, Russell AP, Seydoux J, Montani JP, Buchala A et al. Skeletal muscle insulin and leptin resistance during early catch-up fat driven by suppressed thermogenesis: a role for impaired PI3K and AMPK signalling. Int J Obes Rel Metab Disord 2006 (Abstract) 30: S70.
Crescenzo R, Lionetti L, Mollica MP, Ferraro M, D'Andrea E, Mainieri D et al. Altered skeletal muscle subsarcolemmal mitochondrial compartment during catch-up fat after caloric restriction. Diabetes 2006; 55: 2286–2293.
Hood D . Plasticity in skeletal, cardiac, and smooth muscle: contractile activity-induced mitochondrial biogenesis in skeletal muscle. J Appl Physiol 2001; 90: 1137–1157.
Dulloo AG, Gubler M, Montani JP, Seydoux J, Solinas G . Substrate cycling between de novo lipogenesis and lipid oxidation: a thermogenic mechanism against skeletal muscle lipotoxicity and glucolipotoxicity. Int J Obes Relat Metab Disord 2004; 28 (Suppl 4): S29–S37.
Mainieri D, Summermatter S, Rusconi S, Russell AP, Seydoux J, Montani JP et al. A role for stearoyl-CoA desaturase in control of thermogenesis. FASEB J 2006; 20: 1751–1753.
Voss MD, Beha A, Tennagels N, Tschank G, Herling AW, Quint M et al. Gene expression profiling in skeletal muscle of Zucker diabetic fatty rats: implications for a role of stearoyl-CoA desaturase 1 in insulin resistance. Diabetologia 2005; 48: 2622–2630.
Rahman SM, Dobrzyn A, Dobrzyn P, Lee SH, Miyazaki M, Ntambi JM . Stearoyl-CoA desaturase 1 deficiency elevates insulin-signaling components and down-regulates protein-tyrosine phosphatase 1B in muscle. Proc Natl Acad Sci USA 2003; 100: 11110–11115.
Dobrzyn A, Dobrzyn P, Lee SH, Miyazaki M, Cohen P, Asilmaz E et al. Stearoyl-CoA desaturase-1 deficiency reduces ceramide synthesis by downregulating serine palmitoyltransferase and increasing beta-oxidation in skeletal muscle. Am J Physiol 2005; 288: E599–E607.
Kensara OA, Wootton SA, Phillips DI, Patel M, Hoffman DJ, Jackson AA et al. Substrate-energy metabolism and metabolic risk for cardiovascular disease in relation to fetal growth and adult body composition. Am J Physiol 2006; 291: E365–E371.
Acknowledgements
This work is funded by the Swiss National Science Research Foundation (Grants # 3200-B0-102156 and # 3200B0-113634).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Dulloo, A., Jacquet, J., Seydoux, J. et al. The thrifty ‘catch-up fat’ phenotype: its impact on insulin sensitivity during growth trajectories to obesity and metabolic syndrome. Int J Obes 30 (Suppl 4), S23–S35 (2006). https://doi.org/10.1038/sj.ijo.0803516
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.ijo.0803516
Keywords
This article is cited by
-
Cord blood epigenome-wide meta-analysis in six European-based child cohorts identifies signatures linked to rapid weight growth
BMC Medicine (2023)
-
Branched-chain amino acid supplementation does not enhance lean tissue accretion in low birth weight neonatal pigs, despite lower Sestrin2 expression in skeletal muscle
Amino Acids (2023)
-
Maternal weight, blood lipids, and the offspring weight trajectories during infancy and early childhood in twin pregnancies
World Journal of Pediatrics (2023)
-
Exclusive human milk feeding and prevalence of early adiposity rebound in ELBW infants: a retrospective cohort study
European Journal of Pediatrics (2023)
-
Complementary feeding in preterm infants: a position paper by Italian neonatal, paediatric and paediatric gastroenterology joint societies
Italian Journal of Pediatrics (2022)
