
Abstract
Cohort and case-control data have suggested an association between low to moderate alcohol consumption and decreased risk of ischemic heart disease (IHD), yet results from Mendelian randomization (MR) studies designed to reduce bias have shown either no or a harmful association. Here we conducted an updated systematic review and re-evaluated existing cohort, case-control, and MR data using the burden of proof meta-analytical framework. Cohort and case-control data show low to moderate alcohol consumption is associated with decreased IHD risk – specifically, intake is inversely related to IHD and myocardial infarction morbidity in both sexes and IHD mortality in males – while pooled MR data show no association, confirming that self-reported versus genetically predicted alcohol use data yield conflicting findings about the alcohol-IHD relationship. Our results highlight the need to advance MR methodologies and emulate randomized trials using large observational databases to obtain more definitive answers to this critical public health question.
Similar content being viewed by others


Introduction
It is well known that alcohol consumption increases the risk of morbidity and mortality due to many health conditions1,2, with even low levels of consumption increasing the risk for some cancers3,4. In contrast, a large body of research has suggested that low to moderate alcohol intake – compared to no consumption – is associated with a decreased risk of ischemic heart disease (IHD). This has led to substantial epidemiologic and public health interest in the alcohol-IHD relationship5, particularly given the high prevalence of alcohol consumption6 and the global burden of IHD7.
Extensive evidence from experimental studies that vary short-term alcohol exposure suggests that average levels of alcohol intake positively affect biomarkers such as apolipoprotein A1, adiponectin, and fibrinogen levels that lower the risk of IHD8. In contrast, heavy episodic drinking (HED) may have an adverse effect on IHD by affecting blood lipids, promoting coagulation and thus thrombosis risk, and increasing blood pressure9. With effects likely to vary materially by patterns of drinking, alcohol consumption must be considered a multidimensional factor impacting IHD outcomes.
A recent meta-analysis of the alcohol-IHD relationship using individual participant data from 83 observational studies4 found, among current drinkers, that – relative to drinking less than 50 g/week – any consumption above this level was associated with a lower risk of myocardial infarction (MI) incidence and consumption between >50 and <100 g/week was associated with lower risk of MI mortality. When evaluating other subtypes of IHD excluding MI, the researchers found that consumption between >100 and <250 g/week was associated with a decreased risk of IHD incidence, whereas consumption greater than 350 g/week was associated with an increased risk of IHD mortality. Roerecke and Rehm further observed that low to moderate drinking was not associated with reduced IHD risk when accompanied by occasional HED10.
The cohort studies and case-control studies (hereafter referred to as ‘conventional observational studies’) used in these meta-analyses are known to be subject to various types of bias when used to estimate causal relationships11. First, neglecting to separate lifetime abstainers from former drinkers, some of whom may have quit due to developing preclinical symptoms (sometimes labeled ‘sick quitters’12,13), and to account for drinkers who reduce their intake as a result of such symptoms may introduce reverse causation bias13. That is, the risk of IHD in, for example, individuals with low to moderate alcohol consumption may be lower when compared to IHD risk in sick quitters, not necessarily because intake at this level causes a reduction in risk but because sick quitters are at higher risk of IHD. Second, estimates can be biased because of measurement error in alcohol exposure resulting from inaccurate reporting, random fluctuation in consumption over time (random error), or intentional misreporting of consumption due, for example, to social desirability effects14 (systematic error). Third, residual confounding may bias estimates if confounders of the alcohol-IHD relationship, such as diet or physical activity, have not been measured accurately (e.g., only via a self-report questionnaire) or accounted for. Fourth, because alcohol intake is a time-varying exposure, time-varying confounding affected by prior exposure must be accounted for15. To date, only one study that used a marginal structural model to appropriately adjust for time-varying confounding found no association between alcohol consumption and MI risk16. Lastly, if exposure to a risk factor, such as alcohol consumption, did not happen at random – even if all known confounders of the relationship between alcohol and IHD were perfectly measured and accounted for – the potential for unmeasured confounders persists and may bias estimates11.
In recent years, the analytic method of Mendelian randomization (MR) has been widely adopted to quantify the causal effects of risk factors on health outcomes17,18,19. MR uses single nucleotide polymorphisms (SNPs) as instrumental variables (IVs) for the exposure of interest. A valid IV should fulfill the following three assumptions: it must be associated with the risk factor (relevance assumption); there must be no common causes of the IV and the outcome (independence assumption); and the IV must affect the outcome only through the exposure (exclusion restriction or ‘no horizontal pleiotropy’ assumption)20,21. If all three assumptions are fulfilled, estimates derived from MR are presumed to represent causal effects22. Several MR studies have quantified the association between alcohol consumption and cardiovascular disease23, including IHD, using genes known to impact alcohol metabolism (e.g., ADH1B/C and ALDH224) or SNP combinations from genome-wide association studies25. In contrast to the inverse associations found in conventional observational studies, MR studies have found either no association or a harmful relationship between alcohol consumption and IHD26,27,28,29,30,31.
To advance the knowledge base underlying our understanding of this major health issue – critical given the worldwide ubiquity of alcohol use and of IHD – there is a need to systematically review and critically re-evaluate all available evidence on the relationship between alcohol consumption and IHD risk from both conventional observational and MR studies.
The burden of proof approach, developed by Zheng et al.32, is a six-step meta-analysis framework that provides conservative estimates and interpretations of risk-outcome relationships. The approach systematically tests and adjusts for common sources of bias defined according to the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) criteria: representativeness of the study population, exposure assessment, outcome ascertainment, reverse causation, control for confounding, and selection bias. The key statistical tool to implement the approach is MR-BRT (meta-regression—Bayesian, regularized, trimmed33), a flexible meta-regression tool that does not impose a log-linear relationship between the risk and outcome, but instead uses a spline ensemble to model non-linear relationships. MR-BRT also algorithmically detects and trims outliers in the input data, takes into account different reference and alternative exposure intervals in the data, and incorporates unexplained between-study heterogeneity in the uncertainty surrounding the mean relative risk (RR) curve (henceforth ‘risk curve’). For those risk-outcome relationships that meet the condition of statistical significance using conventionally estimated uncertainty intervals (i.e., without incorporating unexplained between-study heterogeneity), the burden of proof risk function (BPRF) is derived by calculating the 5th (if harmful) or 95th (if protective) quantile risk curve – inclusive of between-study heterogeneity – closest to the log RR of 0. The resulting BPRF is a conservative interpretation of the risk-outcome relationship based on all available evidence. The BPRF represents the smallest level of excess risk for a harmful risk factor or reduced risk for a protective risk factor that is consistent with the data, accounting for between-study heterogeneity. To quantify the strength of the evidence for the alcohol-IHD relationship, the BPRF can be summarized in a single metric, the risk-outcome score (ROS). The ROS is defined as the signed value of the average log RR of the BPRF across the 15th to 85th percentiles of alcohol consumption levels observed across available studies. The larger a positive ROS value, the stronger the alcohol-IHD association. For ease of interpretation, the ROS is converted into a star rating from one to five. A one-star rating (ROS < 0) indicates a weak alcohol-IHD relationship, and a five-star rating (ROS > 0.62) indicates a large effect size and strong evidence. Publication and reporting bias are evaluated with Egger’s regression and by visual inspection with funnel plots34. Further conceptual and technical details of the burden of proof approach are described in detail elsewhere32.
Using the burden of proof approach, we systematically re-evaluate all available eligible evidence from cohort, case-control, and MR studies published between 1970 and 2021 to conservatively quantify the dose-response relationship between alcohol consumption and IHD risk, calculated relative to risk at zero alcohol intake (i.e., current non-drinking, including lifetime abstinence or former use). We pool the evidence from all conventional observational studies combined, as well as individually for all three study designs, to estimate mean IHD risk curves. Based on patterns of results established by previous meta-analyses4,35, we also use data from conventional observational studies to estimate risk curves by IHD endpoint (morbidity or mortality) and further by sex, in addition to estimating risk curves for MI overall and by endpoint. We follow PRISMA (Preferred Reporting Items for Systematic Reviews and Meta-Analyses) guidelines36 through all stages of this study (Supplementary Information section 1, Fig. S1 and Tables S1 and S2) and comply with GATHER (Guidelines on Accurate and Transparent Health Estimates Reporting) recommendations37 (Supplementary Information section 2, Table S3). The main findings and research implications of this work are summarized in Table 1.
Results
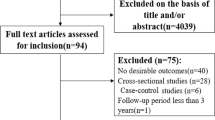
We updated the systematic review on the dose-response relationship between alcohol consumption and IHD previously conducted for the Global Burden of Diseases, Injuries, and Risk Factors Study (GBD) 20201. Of 4826 records identified in our updated systematic review (4769 from databases/registers and 57 by citation search and known literature), 11 were eligible based on our inclusion criteria and were included. In total, combined with the results of the previous systematic reviews1,38, information from 95 cohort studies26,27,29,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130, 27 case-control studies131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157, and five MR studies26,27,28,29,31 was included in our meta-analysis (see Supplementary Information section 1, Fig. S1, for the PRISMA diagram). Details on the extracted effect sizes, the design of each included study, underlying data sources, number of participants, duration of follow-up, number of cases and controls, and bias covariates that were evaluated and potentially adjusted for can be found in the Supplementary Information Sections 4, 5, and 6.
Table 2 summarizes key metrics of each risk curve modeled, including estimates of mean RR and 95% UI (inclusive of between-study heterogeneity) at select alcohol exposure levels, the exposure level and RR and 95% UI at the nadir (i.e., lowest RR), the 85th percentile of exposure observed in the data and its corresponding RR and 95% UI, the BPRF averaged at the 15th and 85th percentile of exposure, the average excess risk or risk reduction according to the exposure-averaged BPRF, the ROS, the associated star rating, the potential presence of publication or reporting bias, and the number of studies included.
We found large variation in the association between alcohol consumption and IHD by study design. When we pooled the results of cohort and case-control studies, we observed an inverse association between alcohol at average consumption levels and IHD risk; that is, drinking average levels of alcohol was associated with a reduced IHD risk relative to drinking no alcohol. In contrast, we did not find a statistically significant association between alcohol consumption and IHD risk when pooling results from MR studies. When we subset the conventional observational studies to those reporting on IHD by endpoint, we found no association between alcohol consumption and IHD morbidity or mortality due to large unexplained heterogeneity between studies. When we further subset those studies that reported effect size estimates by sex, we found that average alcohol consumption levels were inversely associated with IHD morbidity in males and in females, and with IHD mortality in males but not in females. When we analyzed only the studies that reported on MI, we found significant inverse associations between average consumption levels and MI overall and with MI morbidity. Visualizations of the risk curves for morbidity and mortality of IHD and MI are provided in Supplementary Information Section 9 (Figs. S2a–c, S3a–c, and S4a–c). Among all modeled risk curves for which a BPRF was calculated, the ROS ranged from −0.40 for MI mortality to 0.20 for MI morbidity. In the Supplementary Information, we also provide details on the RR and 95% UIs with and without between-study heterogeneity associated with each 10 g/day increase in consumption for each risk curve (Table S10), the parameter specifications of the model (Tables S11 and S12), and each risk curve from the main analysis estimated without trimming 10% of the data (Fig. S5a–l and Table S13).
Risk curve derived from conventional observational study data
The mean risk curve and 95% UI were first estimated by combining all evidence from eligible cohort and case-control studies that quantified the association between alcohol consumption and IHD risk. In total, information from 95 cohort studies and 27 case-control studies combining data from 7,059,652 participants were included. In total, 243,357 IHD events were recorded. Thirty-seven studies quantified the association between alcohol consumption and IHD morbidity only, and 44 studies evaluated only IHD mortality. The estimated alcohol-IHD association was adjusted for sex and age in all but one study. Seventy-five studies adjusted the effect sizes for sex, age, smoking, and at least four other covariates. We adjusted our risk curve for whether the study sample was under or over 50 years of age, whether the study outcome was consistent with the definition of IHD (according to the International Classification of Diseases [ICD]−9: 410-414; and ICD-10: I20-I25) or related to specified subtypes of IHD, whether the outcome was ascertained by self-report only or by at least one other measurement method, whether the study accounted for risk for reverse causation, whether the reference group was non-drinkers (including lifetime abstainers and former drinkers), and whether effect sizes were adjusted (1) for sex, age, smoking, and at least four other variables, (2) for apolipoprotein A1, and (3) for cholesterol, as these bias covariates were identified as significant by our algorithm.
Pooling all data from cohort and case-control studies, we found that alcohol consumption was inversely associated with IHD risk (Fig. 1). The risk curve was J-shaped – without crossing the null RR of 1 at high exposure levels – with a nadir of 0.69 (95% UI: 0.48–1.01) at 23 g/day. This means that compared to individuals who do not drink alcohol, the risk of IHD significantly decreases with increasing consumption up to 23 g/day, followed by a risk reduction that becomes less pronounced. The average BPRF calculated between 0 and 45 g/day of alcohol intake (the 15th and 85th percentiles of the exposure range observed in the data) was 0.96. Thus, when between-study heterogeneity is accounted for, a conservative interpretation of the evidence suggests drinking alcohol across the average intake range is associated with an average decrease in the risk of IHD of at least 4% compared to drinking no alcohol. This corresponds to a ROS of 0.04 and a star rating of two, which suggests that the association – on the basis of the available evidence – is weak. Although we algorithmically identified and trimmed 10% of the data to remove outliers, Egger’s regression and visual inspection of the funnel plot still indicated potential publication or reporting bias.

The panels show the log(relative risk) function, the relative risk function, and a modified funnel plot showing the residuals (relative to 0) on the x-axis and the estimated standard error that includes the reported standard error and between-study heterogeneity on the y-axis. RR relative risk, UI uncertainty interval. Source data are provided as a Source Data file.
Risk curve derived from case-control study data
Next, we estimated the mean risk curve and 95% UI for the relationship between alcohol consumption and IHD by subsetting the data to case-control studies only. We included a total of 27 case-control studies (including one nested case-control study) with data from 60,914 participants involving 16,892 IHD cases from Europe (n = 15), North America (n = 6), Asia (n = 4), and Oceania (n = 2). Effect sizes were adjusted for sex and age in most studies (n = 25). Seventeen of these studies further adjusted for smoking and at least four other covariates. The majority of case-control studies accounted for the risk of reverse causation (n = 25). We did not adjust our risk curve for bias covariates, as our algorithm did not identify any as significant.
Evaluating only data from case-control studies, we observed a J-shaped relationship between alcohol consumption and IHD risk, with a nadir of 0.65 (0.50–0.85) at 23 g/day (Fig. 2). The inverse association between alcohol consumption and IHD risk reversed at an intake level of 61 g/day. In other words, alcohol consumption between >0 and 60 g/day was associated with a lower risk compared to no consumption, while consumption at higher levels was associated with increased IHD risk. However, the curve above this level is flat, implying that the association between alcohol and increased IHD risk is the same between 61 and 100 g/day, relative to not drinking any alcohol. The BPRF averaged across the exposure range between the 15th and 85th percentiles, or 0–45 g/day, was 0.87, which translates to a 13% average reduction in IHD risk across the average range of consumption. This corresponds to a ROS of 0.14 and a three-star rating. After trimming 10% of the data, no potential publication or reporting bias was found.

The panels show the log(relative risk) function, the relative risk function, and a modified funnel plot showing the residuals (relative to 0) on the x-axis and the estimated standard deviation that includes the reported standard deviation and between-study heterogeneity on the y-axis. RR relative risk, UI uncertainty interval. Source data are provided as a Source Data file.
Risk curve derived from cohort study data
We also estimated the mean risk curve and 95% UI for the relationship between alcohol consumption and IHD using only data from cohort studies. In total, 95 cohort studies – of which one was a retrospective cohort study – with data from 6,998,738 participants were included. Overall, 226,465 IHD events were recorded. Most data were from Europe (n = 43) and North America (n = 33), while a small number of studies were conducted in Asia (n = 14), Oceania (n = 3), and South America (n = 2). The majority of studies adjusted effect sizes for sex and age (n = 76). Fifty-seven of these studies also adjusted for smoking and at least four other covariates. Out of all cohort studies included, 88 accounted for the risk of reverse causation. We adjusted our risk curve for whether the study outcome was consistent with the definition of IHD or related to specified subtypes of IHD, and whether effect sizes were adjusted for apolipoprotein A1, as these bias covariates were identified as significant by our algorithm.
When only data from cohort studies were evaluated, we found a J-shaped relationship between alcohol consumption and IHD risk that did not cross the null RR of 1 at high exposure levels, with a nadir of 0.69 (0.47–1.01) at 23 g/day (Fig. 3). The shape of the risk curve was almost identical to the curve estimated with all conventional observational studies (i.e., cohort and case-control studies combined). When we calculated the average BPRF of 0.95 between the 15th and 85th percentiles of observed alcohol exposure (0–50 g/day), we found that alcohol consumption across the average intake range was associated with an average reduction in IHD risk of at least 5%. This corresponds to a ROS of 0.05 and a two-star rating. We identified potential publication or reporting bias after 10% of the data were trimmed.

The panels show the log(relative risk) function, the relative risk function, and a modified funnel plot showing the residuals (relative to 0) on the x-axis and the estimated standard deviation that includes the reported standard deviation and between-study heterogeneity on the y-axis. RR relative risk, UI uncertainty interval. Source data are provided as a Source Data file.
Risk curve derived from Mendelian randomization study data
Lastly, we pooled evidence on the relationship between genetically predicted alcohol consumption and IHD risk from MR studies. Four MR studies were considered eligible for inclusion in our main analysis, with data from 559,708 participants from China (n = 2), the Republic of Korea (n = 1), and the United Kingdom (n = 1). Overall, 22,134 IHD events were recorded. Three studies used the rs671 ALDH2 genotype found in Asian populations, one study additionally used the rs1229984 ADH1B variant, and one study used the rs1229984 ADH1B Arg47His variant and a combination of 25 SNPs as IVs. All studies used the two-stage least squares (2SLS) method to estimate the association, and one study additionally applied the inverse-variance-weighted (IVW) method and multivariable MR (MVMR). For the study that used multiple methods to estimate effect sizes, we used the 2SLS estimates for our main analysis. Further details on the included studies are provided in Supplementary Information section 4 (Table S6). Due to limited input data, we elected not to trim 10% of the observations. We adjusted our risk curve for whether the endpoint of the study outcome was mortality and whether the associations were adjusted for sex and/or age, as these bias covariates were identified as significant by our algorithm.
We did not find any significant association between genetically predicted alcohol consumption and IHD risk using data from MR studies (Fig. 4). No potential publication or reporting bias was detected.

The panels show the log(relative risk) function, the relative risk function, and a modified funnel plot showing the residuals (relative to 0) on the x-axis and the estimated standard deviation that includes the reported standard deviation and between-study heterogeneity on the y-axis. RR relative risk, UI uncertainty interval. Source data are provided as a Source Data file.
As sensitivity analyses, we modeled risk curves with effect sizes estimated from data generated by Lankester et al.28 using IVW and MVMR methods. We also used effect sizes from Biddinger et al.31, obtained using non-linear MR with the residual method, instead of those from Lankester et al.28 in our main model (both were estimated with UK Biobank data) to estimate a risk curve. Again, we did not find a significant association between genetically predicted alcohol consumption and IHD risk (see Supplementary Information Section 10, Fig. S6a–c and Table S14). To test for consistency with the risk curve we estimated using all included cohort studies, we also pooled the conventionally estimated effect sizes provided in the four MR studies. We did not observe an association between alcohol consumption and IHD risk due to large unexplained heterogeneity between studies (see Supplementary Information Section 10, Fig. S7, and Table S14). Lastly, we pooled cohort studies that included data from China, the Republic of Korea, and the United Kingdom to account for potential geographic influences. Again, we did not find a significant association between alcohol consumption and IHD risk (see Supplementary Information Section 10, Fig. S8, and Table S14).
Discussion
Conventional observational and MR studies published to date provide conflicting estimates of the relationship between alcohol consumption and IHD. We conducted an updated systematic review and conservatively re-evaluated existing evidence on the alcohol-IHD relationship using the burden of proof approach. We synthesized evidence from cohort and case-control studies combined and separately and from MR studies to assess the dose-response relationship between alcohol consumption and IHD risk and to compare results across different study designs. It is anticipated that the present synthesis of evidence will be incorporated into upcoming iterations of GBD.
Our estimate of the association between genetically predicted alcohol consumption and IHD runs counter to our estimates from the self-report data and those of other previous meta-analyses4,35,158 that pooled conventional observational studies. Based on the conservative burden of proof interpretation of the data, our results suggested an inverse association between alcohol and IHD when all conventional observational studies were pooled (alcohol intake was associated with a reduction in IHD risk by an average of at least 4% across average consumption levels; two-star rating). In evaluating only cohort studies, we again found an inverse association between alcohol consumption and IHD (alcohol intake was associated with a reduction in IHD risk by an average of at least 5% at average consumption levels; two-star rating). In contrast, when we pooled only case-control studies, we estimated that average levels of alcohol consumption were associated with at least a 13% average decrease in IHD risk (three-star rating), but the inverse association reversed when consumption exceeded 60 g/day, suggesting that alcohol above this level is associated with a slight increase in IHD risk. Our analysis of the available evidence from MR studies showed no association between genetically predicted alcohol consumption and IHD.
Various potential biases and differences in study designs may have contributed to the conflicting findings. In our introduction, we summarized important sources of bias in conventional observational studies of the association between alcohol consumption and IHD. Of greatest concern are residual and unmeasured confounding and reverse causation, the effects of which are difficult to eliminate in conventional observational studies. By using SNPs within an IV approach to predict exposure, MR – in theory – eliminates these sources of bias and allows for more robust estimates of causal effects. Bias may still occur, however, when using MR to estimate the association between alcohol and IHD159,160. There is always the risk of horizontal pleiotropy in MR – that is, the genetic variant may affect the outcome via pathways other than exposure161. The IV assumption of exclusion restriction is, for example, violated if only a single measurement of alcohol consumption is used in MR162; because alcohol consumption varies over the life course, the gene directly impacts IHD through intake at time points other than that used in the MR analysis. To date, MR studies have not succeeded in separately capturing the multidimensional effects of alcohol intake on IHD risk (i.e., effects of average alcohol consumption measured through frequency-quantity, in addition to the effects of HED)159 because the genes used to date only target average alcohol consumption that encompasses intake both at average consumption levels and HED. In other words, the instruments used are not able to separate out the individual effects of these two different dimensions of alcohol consumption on IHD risk using MR. Moreover, reverse causation may occur through cross-generational effects160,163, as the same genetic variants predispose both the individual and at least one of his or her parents to (increased) alcohol consumption. In this situation, IHD risk could be associated with the parents’ genetically predicted alcohol consumption and not with the individual’s own consumption. None of the MR studies included accounted for cross-generational effects, which possibly introduced bias in the effect estimates. It is important to note that bias by ancestry might also occur in conventional observational studies164. In summary, estimates of the alcohol-IHD association are prone to bias in all three study designs, limiting inferences of causation.
The large difference in the number of available MR versus conventional observational studies, the substantially divergent results derived from the different study types, and the rapidly developing field of MR clearly argue for further investigation of MR as a means to quantify the association between alcohol consumption and IHD risk. Future studies should investigate non-linearity in the relationship using non-linear MR methods. The residual method, commonly applied in non-linear MR studies such as Biddinger et al.31, assumes a constant, linear relationship between the genetic IV and the exposure in the study population; a strong assumption that may result in biased estimates and inflated type I error rates if the relationship varies by population strata165. However, by log-transforming the exposure, the relationships between the genetic IV and the exposure as expressed on a logarithmic scale may be more homogeneous across strata, possibly reducing the bias effect of violating the assumption of a constant, linear relationship. Alternatively, or in conjunction, the recently developed doubly ranked method, which obviates the need for this assumption, could be used166. Since methodology for non-linear MR is an active field of study167, potential limitations of currently available methods should be acknowledged and latest guidelines be followed168. Future MR studies should further (i) employ sensitivity analyses such as the MR weighted median method169 to relax the exclusion restriction assumption that may be violated, as well as applying other methods such as the MR-Egger intercept test; (ii) use methods such as g-estimation of structural mean models162 to adequately account for temporal variation in alcohol consumption in MR, and (iii) attempt to disaggregate the effects of alcohol on IHD by dimension in MR, potentially through the use of MVMR164. General recommendations to overcome common MR limitations are described in greater detail elsewhere159,163,170,171 and should be carefully considered. With respect to prospective cohort studies used to assess the alcohol-IHD relationship, they should, at a minimum: (i) adjust the association between alcohol consumption and IHD for all potential confounders identified, for example, using a causal directed acyclic graph, and (ii) account for reverse causation introduced by sick quitters and by drinkers who changed their consumption. If possible, they should also (iii) use alcohol biomarkers as objective measures of alcohol consumption instead of or in addition to self-reported consumption to reduce bias through measurement error, (iv) investigate the association between IHD and HED, in addition to average alcohol consumption, and (v) when multiple measures of alcohol consumption and potential confounders are available over time, use g-methods to reduce bias through confounding as fully as possible within the limitations of the study design. However, some bias – due, for instance, to unmeasured confounding in conventional observational and to horizontal pleiotropy in MR studies – is likely inevitable, and the interpretation of estimates should be appropriately cautious, in accordance with the methods used in the study.
With the introduction of the Moderate Alcohol and Cardiovascular Health Trial (MACH15)172, randomized controlled trials (RCTs) have been revisited as a way to study the long-term effects of low to moderate alcohol consumption on cardiovascular disease, including IHD. In 2018, soon after the initiation of MACH15, the National Institutes of Health terminated funding173, reportedly due to concerns about study design and irregularities in the development of funding opportunities174. Although MACH15 was terminated, its initiation represented a previously rarely considered step toward investigating the alcohol-IHD relationship using an RCT175. However, while the insights from an RCT are likely to be invaluable, the implementation is fraught with potential issues. Due to the growing number of studies suggesting increased disease risk, including cancer3,4, associated with alcohol use even at very low levels176, the use of RCTs to study alcohol consumption is ethically questionable177. A less charged approach could include the emulation of target trials178 using existing observational data (e.g., from large-scale prospective cohort studies such as the UK Biobank179, Atherosclerosis Risk in Communities Study180, or the Framingham Heart Study181) in lieu of real trials to gather evidence on the potential cardiovascular effects of alcohol. Trials like MACH15 can be emulated, following the proposed trial protocols as closely as the observational dataset used for the analysis allows. Safety and ethical concerns, such as those related to eligibility criteria, initiation/increase in consumption, and limited follow-up duration, will be eliminated because the data will have already been collected. This framework allows for hypothetical trials investigating ethically challenging or even untenable questions, such as the long-term effects of heavy (episodic) drinking on IHD risk, to be emulated and inferences to broader populations drawn.
There are several limitations that must be considered when interpreting our findings. First, record screening for our systematic review was not conducted in a double-blinded fashion. Second, we did not have sufficient evidence to estimate and examine potential differential associations of alcohol consumption with IHD risk by beverage type or with MI endpoints by sex. Third, despite using a flexible meta-regression tool that overcame several limitations common to meta-analyses, the results of our meta-analysis were only as good as the quality of the studies included. We were able, however, to address the issue of varying quality of input data by adjusting for bias covariates that corresponded to core study characteristics in our analyses. Fourth, because we were only able to include one-sample MR studies that captured genetically predicted alcohol consumption, statistical power may be lower than would have been possible with the inclusion of two-sample MR studies, and studies that directly estimated gene-IHD associations were not considered23. Finally, we were not able to account for participants’ HED status when pooling effect size estimates from conventional observational studies. Given established differences in IHD risk for drinkers with and without HED35 and the fact that more than one in three drinkers reports HED6, we would expect that the decreased average risk we found at moderate levels of alcohol consumption would be attenuated (i.e., approach the IHD risk of non-drinkers) if the presence of HED was taken into account.
Using the burden of proof approach32, we conservatively re-evaluated the dose-response relationship between alcohol consumption and IHD risk based on existing cohort, case-control, and MR data. Consistent with previous meta-analyses, we found that alcohol at average consumption levels was inversely associated with IHD when we pooled conventional observational studies. This finding was supported when aggregating: (i) all studies, (ii) only cohort studies, (iii) only case-control studies, (iv) studies examining IHD morbidity in females and males, (v) studies examining IHD mortality in males, and (vi) studies examining MI morbidity. In contrast, we found no association between genetically predicted alcohol consumption and IHD risk based on data from MR studies. Our confirmation of the conflicting results derived from self-reported versus genetically predicted alcohol use data highlights the need to advance methodologies that will provide more definitive answers to this critical public health question. Given the limitations of randomized trials, we advocate using advanced MR techniques and emulating target trials using observational data to generate more conclusive evidence on the long-term effects of alcohol consumption on IHD risk.
Methods
This study was approved by the University of Washington IRB Committee (study #9060).
Overview
The burden of proof approach is a six-step framework for conducting meta-analysis32: (1) data from published studies that quantified the dose-response relationship between alcohol consumption and ischemic heart disease (IHD) risk were systematically identified and obtained; (2) the shape of the mean relative risk (RR) curve (henceforth ‘risk curve’) and associated uncertainty was estimated using a quadratic spline and algorithmic trimming of outliers; (3) the risk curve was tested and adjusted for biases due to study attributes; (4) unexplained between-study heterogeneity was quantified, adjusting for within-study correlation and number of studies included; (5) the evidence for small-study effects was evaluated to identify potential risks of publication or reporting bias; and (6) the burden of proof risk function (BPRF) – a conservative interpretation of the average risk across the exposure range found in the data – was estimated relative to IHD risk at zero alcohol intake. The BPRF was converted to a risk-outcome score (ROS) that was mapped to a star rating from one to five to provide an intuitive interpretation of the magnitude and direction of the dose-response relationship between alcohol consumption and IHD risk.
We calculated the mean RR and 95% uncertainty intervals (UIs) for IHD associated with levels of alcohol consumption separately with all evidence available from conventional observational studies and from Mendelian randomization (MR) studies. For the risk curves that met the condition of statistical significance when the conventional 95% UI that does not include unexplained between-study heterogeneity was evaluated, we calculated the BPRF, ROS, and star rating. Based on input data from conventional observational studies, we also estimated these metrics by study design (cohort studies, case-control studies), and by IHD endpoint (morbidity, mortality) for both sexes (females, males) and sex-specific. For sex-stratified analyses, we only considered studies that reported effect sizes for both females and males to allow direct comparison of IHD risk across different exposure levels; however, we did not collect information about the method each study used to determine sex. We also estimated risk curves for myocardial infarction (MI), overall and by endpoint, using data from conventional observational studies. As a comparison, we also estimated each risk curve without trimming 10% of the input data. We did not consider MI as an outcome or disaggregate findings by sex or endpoint for MR studies due to insufficient data.
With respect to MR studies, several statistical methods are typically used to estimate the associations between genetically predicted exposure and health outcomes (e.g., two-stage least squares [2SLS], inverse-variance-weighted [IVW], multivariable Mendelian randomization [MVMR]). For our main analysis synthesizing evidence from MR studies, we included the reported effect sizes estimated using 2SLS if a study applied multiple methods because this method was common to all included studies. In sensitivity analyses, we used the effect sizes obtained by other MR methods (i.e., IVW, MVMR, and non-linear MR) and estimated the mean risk curve and uncertainty. We also pooled conventionally estimated effect sizes from MR studies to allow comparison with the risk curve estimated with cohort studies. Due to limited input data from MR studies, we elected not to trim 10% of the observations. Furthermore, we estimated the risk curve from cohort studies with data from countries that corresponded to those included in MR studies (China, the Republic of Korea, and the United Kingdom). Due to a lack of data, we were unable to estimate a risk curve from case-control studies in these geographic regions.
Conducting the systematic review
In step one of the burden of proof approach, data for the dose-response relationship between alcohol consumption and IHD risk were systematically identified, reviewed, and extracted. We updated a previously published systematic review1 in PubMed that identified all studies evaluating the dose-response relationship between alcohol consumption and risk of IHD morbidity or mortality from January 1, 1970, to December 31, 2019. In our update, we additionally considered all studies up to and including December 31, 2021, for eligibility. We searched articles in PubMed on March 21, 2022, with the following search string: (alcoholic beverage[MeSH Terms] OR drinking behavior[MeSH Terms] OR “alcohol”[Title/Abstract]) AND (Coronary Artery Disease[Mesh] OR Myocardial Ischemia[Mesh] OR atherosclerosis[Mesh] OR Coronary Artery Disease[TiAb] OR Myocardial Ischemia[TiAb] OR cardiac ischemia[TiAb] OR silent ischemia[TiAb] OR atherosclerosis Outdent [TiAb] OR Ischemic heart disease[TiAb] OR Ischemic heart disease[TiAb] OR coronary heart disease[TiAb] OR myocardial infarction[TiAb] OR heart attack[TiAb] OR heart infarction[TiAb]) AND (Risk[MeSH Terms] OR Odds Ratio[MeSH Terms] OR “risk”[Title/Abstract] OR “odds ratio”[Title/Abstract] OR “cross-product ratio”[Title/Abstract] OR “hazards ratio”[Title/Abstract] OR “hazard ratio”[Title/Abstract]) AND (“1970/01/01”[PDat]: “2021/12/31”[PDat]) AND (English[LA]) NOT (animals[MeSH Terms] NOT Humans[MeSH Terms]). Studies were eligible for inclusion if they met all of the following criteria: were published between January 1, 1970, and December 31, 2021; were a cohort study, case-control study, or MR study; described an association between alcohol consumption and IHD and reported an effect size estimate (relative risk, hazard ratio, odds ratio); and used a continuous dose as exposure of alcohol consumption. Studies were excluded if they met any of the following criteria: were an aggregate study (meta-analysis or pooled cohort); utilized a study design not designated for inclusion in this analysis: not a cohort study, case-control study, or MR study; were a duplicate study: the underlying sample of the study had also been analyzed elsewhere (we always considered the analysis with the longest follow-up for cohort studies or the most recently published analysis for MR studies); did not report on the exposure of interest: reported on combined exposure of alcohol and drug use or reported alcohol consumption in a non-continuous way; reported an outcome that was not IHD or a composite outcome that included but was not limited to IHD, or outcomes lacked specificity, such as cardiovascular disease or all-cause mortality; were not in English; and were animal studies. All screenings of titles and abstracts of identified records, as well as full texts of potentially eligible studies, and extraction of included studies, were done by a single reviewer (SC or HL) independently. If eligible, studies were extracted for study characteristics, exposure, outcome, adjusted confounders, and effect sizes and their uncertainty. While the previous systematic review only considered cohort and case-control studies, our update also included MR studies. We chose to consider only ‘one-sample’ MR studies, i.e., those in which genes, risk factors, and outcomes were measured in the same participants, and not ‘two-sample’ MR studies in which two different samples were used for the MR analysis so that we could fully capture study-specific information. We re-screened previously identified records for MR studies to consider all published MR studies in the defined time period. We also identified and included in our sensitivity analysis an MR study published in 202231 which used a non-linear MR method to estimate the association between genetically predicted alcohol consumption and IHD. When eligible studies reported both MR and conventionally estimated effect sizes (i.e., for the association between self-reported alcohol consumption and IHD risk), we extracted both. If studies used the same underlying sample and investigated the same outcome in the same strata, we included the study that had the longest follow-up. This did not apply when the same samples were used in conventional observational and MR studies, because they were treated separately when estimating the risk curve of alcohol consumption and IHD. Continuous exposure of alcohol consumption was defined as a frequency-quantity measure182 and converted to g/day. IHD was defined according to the International Classification of Diseases (ICD)−9, 410-414, and ICD-10, I20-I25.
The raw data were extracted with a standardized extraction sheet (see Supplementary Information Section 3, Table S4). For conventional observational studies, when multiple effect sizes were estimated from differently adjusted regression models, we used those estimated with the model reported to be fully adjusted or the one with the most covariates. In the majority of studies, alcohol consumption was categorized based on the exposure range available in the data. If the lower end of a categorical exposure range (e.g., <10 g/day) of an effect size was not specified in the input data, we assumed that this was 0 g/day. If the upper end was not specified (e.g., >20 g/day), it was calculated by multiplying the lower end of the categorical exposure range by 1.5. When the association between alcohol and IHD risk was reported as a linear slope, the average consumption level in the sample was multiplied by the logarithm of the effect size to effectively render it categorical. From the MR study which employed non-linear MR31, five effect sizes and their uncertainty were extracted at equal intervals across the reported range of alcohol exposure using WebPlotDigitizer. To account for the fact that these effect sizes were derived from the same non-linear risk curve, we adjusted the extracted standard errors by multiplying them by the square root of five (i.e., the number of extracted effect sizes). Details on data sources are provided in Supplementary Information Section 4.
Estimating the shape of the risk-outcome relationship
In step two, the shape of the dose-response relationship (i.e., ‘signal’) between alcohol consumption and IHD risk was estimated relative to risk at zero alcohol intake. The meta-regression tool MR-BRT (meta-regression—Bayesian, regularized, trimmed), developed by Zheng et al.33, was used for modeling. To allow for non-linearity, thus relaxing the common assumption of a log-linear relationship, a quadratic spline with two interior knots was used for estimating the risk curve33. We used the following three risk measures from included studies: RRs, odds ratios (ORs), and hazard ratios (HRs). ORs were treated as equivalent to RRs and HRs based on the rare outcome assumption. To counteract the potential influence of knot placement on the shape of the risk curve when using splines, an ensemble model approach was applied. Fifty component models with random knot placements across the exposure domain were computed. These were combined into an ensemble by weighting each model based on model fit and variation (i.e., smoothness of fit to the data). To prevent bias from outliers, a robust likelihood-based approach was applied to trim 10% of the observations. Technical details on estimating the risk curve, use of splines, the trimming procedure, the ensemble model approach, and uncertainty estimation are described elsewhere32,33. Details on the model specifications for each risk curve are provided in Supplementary Information section 8. We first estimated each risk curve without trimming input data to visualize the shape of the curve, which informed knot placement and whether to set a left and/or right linear tail when data were sparse at low or high exposure levels (see Supplementary Information Section 10, Fig. S5a–l).
Testing and adjusting for biases across study designs and characteristics
In step three, the risk curve was tested and adjusted for systematic biases due to study attributes. According to the Grading of Recommendations Assessment, Development, and Evaluation (GRADE) criteria183, the following six bias sources were quantified: representativeness of the study population, exposure assessment, outcome ascertainment, reverse causation, control for confounding, and selection bias. Representativeness was quantified by whether the study sample came from a location that was representative of the underlying geography. Exposure assessment was quantified by whether alcohol consumption was recorded once or more than once in conventional observational studies, or with only one or multiple SNPs in MR studies. Outcome ascertainment was quantified by whether IHD was ascertained by self-report only or by at least one other measurement method. Reverse causation was quantified by whether increased IHD risk among participants who reduced or stopped drinking was accounted for (e.g., by separating former drinkers from lifetime abstainers). Control for confounding factors was quantified by which and how many covariates the effect sizes were adjusted for (i.e., through stratification, matching, weighting, or standardization). Because the most adjusted effect sizes in each study were extracted in the systematic review process and thus may have been adjusted for mediators, we additionally quantified a bias covariate for each of the following potential mediators of the alcohol-IHD relationship: body mass index, blood pressure, cholesterol (excluding high-density lipoprotein cholesterol), fibrinogen, apolipoprotein A1, and adiponectin. Selection bias was quantified by whether study participants were selected and included based on pre-existing disease states. We also quantified and considered as possible bias covariates whether the reference group was non-drinkers, including lifetime abstainers and former drinkers; whether the sample was under or over 50 years of age; whether IHD morbidity, mortality, or both endpoints were used; whether the outcome mapped to IHD or referred only to subtypes of IHD; whether the outcome mapped to MI; and what study design (cohort or case-control) was used when conventional observational studies were pooled. Details on quantified bias covariates for all included studies are provided in Supplementary Information section 5 (Tables S7 and S8). Using a Lasso approach184, the bias covariates were first ranked. They were then included sequentially, based on their ranking, as effect modifiers of the ‘signal’ obtained in step two in a linear meta-regression. Significant bias covariates were included in modeling the final risk curve. Technical details of the Lasso procedure are described elsewhere32.
Quantifying between-study heterogeneity, accounting for heterogeneity, uncertainty, and small number of studies
In step four, the between-study heterogeneity was quantified, accounting for heterogeneity, uncertainty, and small number of studies. In a final linear mixed-effects model, the log RRs were regressed against the ‘signal’ and selected bias covariates, with a random intercept to account for within-study correlation and a study-specific random slope with respect to the ‘signal’ to account for between-study heterogeneity. A Fisher information matrix was used to estimate the uncertainty associated with between-study heterogeneity185 because heterogeneity is easily underestimated or may be zero when only a small number of studies are available. We estimated the mean risk curve with a 95% UI that incorporated between-study heterogeneity, and we additionally estimated a 95% UI without between-study heterogeneity as done in conventional meta-regressions (see Supplementary Information Section 7, Table S10). The 95% UI incorporating between-study heterogeneity was calculated from the posterior uncertainty of the fixed effects (i.e., the ‘signal’ and selected bias covariates) and the 95% quantile of the between-study heterogeneity. The estimate of between-study heterogeneity and the estimate of the uncertainty of the between-study heterogeneity were used to determine the 95% quantile of the between-study heterogeneity. Technical details of quantifying uncertainty of between-study heterogeneity are described elsewhere32.
Evaluating potential for publication or reporting bias
In step five, the potential for publication or reporting bias was evaluated. The trimming algorithm used in step two helps protect against these biases, so risk curves found to have publication or reporting bias using the following methods were derived from data that still had bias even after trimming. Publication or reporting bias was evaluated using Egger’s regression34 and visual inspection using funnel plots. Egger’s regression tested for a significant correlation between residuals of the RR estimates and their standard errors. Funnel plots showed the residuals of the risk curve against their standard errors. We reported publication or reporting bias when identified.
Estimating the burden of proof risk function
In step six, the BPRF was calculated for risk-outcome relationships that were statistically significant when evaluating the conventional 95% UI without between-study heterogeneity. The BPRF is either the 5th (if harmful) or the 95th (if protective) quantile curve inclusive of between-study heterogeneity that is closest to the RR line at 1 (i.e., null); it indicates a conservative estimate of a harmful or protective association at each exposure level, based on the available evidence. The mean risk curve, 95% UIs (with and without between-study heterogeneity), and BPRF (where applicable) are visualized along with included effect sizes using the midpoint of each alternative exposure range (trimmed data points are marked with a red x), with alcohol consumption in g/day on the x-axis and (log)RR on the y-axis.
We calculated the ROS as the average log RR of the BPRF between the 15th and 85th percentiles of alcohol exposure observed in the study data. The ROS summarizes the association of the exposure with the health outcome in a single measure. A higher, positive ROS indicates a larger association, while a negative ROS indicates a weak association. The ROS is identical for protective and harmful risks since it is based on the magnitude of the log RR. For example, a mean log BPRF between the 15th and 85th percentiles of exposure of −0.6 (protective association) and a mean log BPRF of 0.6 (harmful association) would both correspond to a ROS of 0.6. The ROS was then translated into a star rating, representing a conservative interpretation of all available evidence. A star rating of 1 (ROS: <0) indicates weak evidence of an association, a star rating of 2 (ROS: 0–0.14) indicates a >0–15% increased or >0–13% decreased risk, a star rating of 3 (ROS: >0.14–0.41) indicates a >15–50% increased or >13–34% decreased risk, a star rating of 4 (ROS: >0.41–0.62) indicates a >50–85% increased or >34–46% decreased risk, and a star rating of 5 (ROS: >0.62) indicates a >85% increased or >46% decreased risk.
Statistics & reproducibility
The statistical analyses conducted in this study are described above in detail. No statistical method was used to predetermine the sample size. When analyzing data from cohort and case-control studies, we excluded 10% of observations using a trimming algorithm; when analyzing data from MR studies, we did not exclude any observations. As all data used in this meta-analysis were from observational studies, no experiments were conducted, and no randomization or blinding took place.
Reporting summary
Further information on research design is available in the Nature Portfolio Reporting Summary linked to this article.
Data availability
The findings from this study were produced using data extracted from published literature. The relevant studies were identified through a systematic literature review and can all be accessed online as referenced in the current paper26,27,28,29,31,39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55,56,57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84,85,86,87,88,89,90,91,92,93,94,95,96,97,98,99,100,101,102,103,104,105,106,107,108,109,110,111,112,113,114,115,116,117,118,119,120,121,122,123,124,125,126,127,128,129,130,131,132,133,134,135,136,137,138,139,140,141,142,143,144,145,146,147,148,149,150,151,152,153,154,155,156,157. Further details on the relevant studies can be found on the GHDx website (https://ghdx.healthdata.org/record/ihme-data/gbd-alcohol-ihd-bop-risk-outcome-scores). Study characteristics of all relevant studies included in the analyses are also provided in Supplementary Information Section 4 (Tables S5 and S6). The template of the data collection form is provided in Supplementary Information section 3 (Table S4). The source data includes processed data from these studies that underlie our estimates. Source data are provided with this paper.
Code availability
Analyses were carried out using R version 4.0.5 and Python version 3.10.9. All code used for these analyses is publicly available online (https://github.com/ihmeuw-msca/burden-of-proof).
References
Bryazka, D. et al. Population-level risks of alcohol consumption by amount, geography, age, sex, and year: a systematic analysis for the Global Burden of Disease Study 2020. Lancet 400, 185–235 (2022).
World Health Organization. Global Status Report on Alcohol and Health 2018. (World Health Organization, Geneva, Switzerland, 2019).
Bagnardi, V. et al. Alcohol consumption and site-specific cancer risk: a comprehensive dose–response meta-analysis. Br. J. Cancer 112, 580–593 (2015).
Wood, A. M. et al. Risk thresholds for alcohol consumption: combined analysis of individual-participant data for 599 912 current drinkers in 83 prospective studies. Lancet 391, 1513–1523 (2018).
Goel, S., Sharma, A. & Garg, A. Effect of alcohol consumption on cardiovascular health. Curr. Cardiol. Rep. 20, 19 (2018).
Manthey, J. et al. Global alcohol exposure between 1990 and 2017 and forecasts until 2030: a modelling study. Lancet 393, 2493–2502 (2019).
Vos, T. et al. Global burden of 369 diseases and injuries in 204 countries and territories, 1990–2019: a systematic analysis for the Global Burden of Disease Study 2019. Lancet 396, 1204–1222 (2020).
Brien, S. E., Ronksley, P. E., Turner, B. J., Mukamal, K. J. & Ghali, W. A. Effect of alcohol consumption on biological markers associated with risk of coronary heart disease: systematic review and meta-analysis of interventional studies. BMJ 342, d636 (2011).
Rehm, J. et al. The relationship between different dimensions of alcohol use and the burden of disease—an update. Addiction 112, 968–1001 (2017).
Roerecke, M. & Rehm, J. Irregular heavy drinking occasions and risk of ischemic heart disease: a systematic review and meta-analysis. Am. J. Epidemiol. 171, 633–644 (2010).
Hernan, M. A. & Robin, J. M. Causal Inference: What If. (CRC Press, 2023).
Marmot, M. Alcohol and coronary heart disease. Int. J. Epidemiol. 13, 160–167 (1984).
Shaper, A. G., Wannamethee, G. & Walker, M. Alcohol and mortality in British men: explaining the U-shaped curve. Lancet 332, 1267–1273 (1988).
Davis, C. G., Thake, J. & Vilhena, N. Social desirability biases in self-reported alcohol consumption and harms. Addict. Behav. 35, 302–311 (2010).
Mansournia, M. A., Etminan, M., Danaei, G., Kaufman, J. S. & Collins, G. Handling time varying confounding in observational research. BMJ 359, j4587 (2017).
Ilomäki, J. et al. Relationship between alcohol consumption and myocardial infarction among ageing men using a marginal structural model. Eur. J. Public Health 22, 825–830 (2012).
Lawlor, D. A., Harbord, R. M., Sterne, J. A. C., Timpson, N. & Davey Smith, G. Mendelian randomization: using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163 (2008).
Burgess, S., Timpson, N. J., Ebrahim, S. & Davey Smith, G. Mendelian randomization: where are we now and where are we going? Int. J. Epidemiol. 44, 379–388 (2015).
Sleiman, P. M. & Grant, S. F. Mendelian randomization in the era of genomewide association studies. Clin. Chem. 56, 723–728 (2010).
Davies, N. M., Holmes, M. V. & Davey Smith, G. Reading Mendelian randomisation studies: a guide, glossary, and checklist for clinicians. BMJ 362, k601 (2018).
de Leeuw, C., Savage, J., Bucur, I. G., Heskes, T. & Posthuma, D. Understanding the assumptions underlying Mendelian randomization. Eur. J. Hum. Genet. 30, 653–660 (2022).
Sheehan, N. A., Didelez, V., Burton, P. R. & Tobin, M. D. Mendelian randomisation and causal inference in observational epidemiology. PLoS Med. 5, e177 (2008).
Van de Luitgaarden, I. A. et al. Alcohol consumption in relation to cardiovascular diseases and mortality: a systematic review of Mendelian randomization studies. Eur. J. Epidemiol. 1–15 (2021).
Edenberg, H. J. The genetics of alcohol metabolism: role of alcohol dehydrogenase and aldehyde dehydrogenase variants. Alcohol Res. Health 30, 5–13 (2007).
Gelernter, J. et al. Genome-wide association study of maximum habitual alcohol intake in >140,000 U.S. European and African American veterans yields novel risk loci. Biol. Psychiatry 86, 365–376 (2019).
Millwood, I. Y. et al. Conventional and genetic evidence on alcohol and vascular disease aetiology: a prospective study of 500 000 men and women in China. Lancet 393, 1831–1842 (2019).
Au Yeung, S. L. et al. Moderate alcohol use and cardiovascular disease from Mendelian randomization. PLoS ONE 8, e68054 (2013).
Lankester, J., Zanetti, D., Ingelsson, E. & Assimes, T. L. Alcohol use and cardiometabolic risk in the UK Biobank: a Mendelian randomization study. PLoS ONE 16, e0255801 (2021).
Cho, Y. et al. Alcohol intake and cardiovascular risk factors: a Mendelian randomisation study. Sci. Rep. 5, 18422 (2015).
Holmes, M. V. et al. Association between alcohol and cardiovascular disease: Mendelian randomisation analysis based on individual participant data. BMJ 349, g4164 (2014).
Biddinger, K. J. et al. Association of habitual alcohol intake with risk of cardiovascular disease. JAMA Netw. Open 5, e223849–e223849 (2022).
Zheng, P. et al. The Burden of Proof studies: assessing the evidence of risk. Nat. Med. 28, 2038–2044 (2022).
Zheng, P., Barber, R., Sorensen, R. J., Murray, C. J. & Aravkin, A. Y. Trimmed constrained mixed effects models: formulations and algorithms. J. Comput. Graph. Stat. 30, 544–556 (2021).
Egger, M., Smith, G. D., Schneider, M. & Minder, C. Bias in meta-analysis detected by a simple, graphical test. BMJ 315, 629–634 (1997).
Roerecke, M. & Rehm, J. Alcohol consumption, drinking patterns, and ischemic heart disease: a narrative review of meta-analyses and a systematic review and meta-analysis of the impact of heavy drinking occasions on risk for moderate drinkers. BMC Med. 12, 182 (2014).
Page, M. J. et al. The PRISMA 2020 statement: an updated guideline for reporting systematic reviews. Syst. Rev. 10, 89 (2021).
Stevens, G. A. et al. Guidelines for accurate and transparent health estimates reporting: the GATHER statement. PLoS Med. 13, e1002056 (2016).
Griswold, M. G. et al. Alcohol use and burden for 195 countries and territories, 1990–2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 392, 1015–1035 (2018).
Albert, C. M. et al. Moderate alcohol consumption and the risk of sudden cardiac death among US male physicians. Circulation 100, 944–950 (1999).
Arriola, L. et al. Alcohol intake and the risk of coronary heart disease in the Spanish EPIC cohort study. Heart 96, 124–130 (2010).
Bazzano, L. A. et al. Alcohol consumption and risk of coronary heart disease among Chinese men. Int. J. Cardiol. 135, 78–85 (2009).
Bell, S. et al. Association between clinically recorded alcohol consumption and initial presentation of 12 cardiovascular diseases: population based cohort study using linked health records. BMJ 356, j909 (2017).
Bergmann, M. M. et al. The association of pattern of lifetime alcohol use and cause of death in the European prospective investigation into cancer and nutrition (EPIC) study. Int. J. Epidemiol. 42, 1772–1790 (2013).
Beulens, J. W. J. et al. Alcohol consumption and risk for coronary heart disease among men with hypertension. Ann. Intern. Med. 146, 10–19 (2007).
Bobak, M. et al. Alcohol, drinking pattern and all-cause, cardiovascular and alcohol-related mortality in Eastern Europe. Eur. J. Epidemiol. 31, 21–30 (2016).
Boffetta, P. & Garfinkel, L. Alcohol drinking and mortality among men enrolled in an American Cancer Society prospective study. Epidemiology 1, 342–348 (1990).
Britton, A. & Marmot, M. Different measures of alcohol consumption and risk of coronary heart disease and all-cause mortality: 11-year follow-up of the Whitehall II Cohort Study. Addiction 99, 109–116 (2004).
Camargo, C. A. et al. Moderate alcohol consumption and risk for angina pectoris or myocardial infarction in U.S. male physicians. Ann. Intern. Med. 126, 372–375 (1997).
Chang, J. Y., Choi, S. & Park, S. M. Association of change in alcohol consumption with cardiovascular disease and mortality among initial nondrinkers. Sci. Rep. 10, 13419 (2020).
Chiuve, S. E. et al. Light-to-moderate alcohol consumption and risk of sudden cardiac death in women. Heart Rhythm 7, 1374–1380 (2010).
Colditz, G. A. et al. Moderate alcohol and decreased cardiovascular mortality in an elderly cohort. Am. Heart J. 109, 886–889 (1985).
Dai, J., Mukamal, K. J., Krasnow, R. E., Swan, G. E. & Reed, T. Higher usual alcohol consumption was associated with a lower 41-y mortality risk from coronary artery disease in men independent of genetic and common environmental factors: the prospective NHLBI Twin Study. Am. J. Clin. Nutr. 102, 31–39 (2015).
Dam, M. K. et al. Five year change in alcohol intake and risk of breast cancer and coronary heart disease among postmenopausal women: prospective cohort study. BMJ 353, i2314 (2016).
Degerud, E. et al. Associations of binge drinking with the risks of ischemic heart disease and stroke: a study of pooled Norwegian Health Surveys. Am. J. Epidemiol. 190, 1592–1603 (2021).
de Labry, L. O. et al. Alcohol consumption and mortality in an American male population: recovering the U-shaped curve–findings from the normative Aging Study. J. Stud. Alcohol 53, 25–32 (1992).
Doll, R., Peto, R., Boreham, J. & Sutherland, I. Mortality in relation to alcohol consumption: a prospective study among male British doctors. Int. J. Epidemiol. 34, 199–204 (2005).
Dyer, A. R. et al. Alcohol consumption and 17-year mortality in the Chicago Western Electric Company study. Prev. Med. 9, 78–90 (1980).
Ebbert, J. O., Janney, C. A., Sellers, T. A., Folsom, A. R. & Cerhan, J. R. The association of alcohol consumption with coronary heart disease mortality and cancer incidence varies by smoking history. J. Gen. Intern. Med. 20, 14–20 (2005).
Ebrahim, S. et al. Alcohol dehydrogenase type 1 C (ADH1C) variants, alcohol consumption traits, HDL-cholesterol and risk of coronary heart disease in women and men: British Women’s Heart and Health Study and Caerphilly cohorts. Atherosclerosis 196, 871–878 (2008).
Friedman, L. A. & Kimball, A. W. Coronary heart disease mortality and alcohol consumption in Framingham. Am. J. Epidemiol. 124, 481–489 (1986).
Fuchs, F. D. et al. Association between alcoholic beverage consumption and incidence of coronary heart disease in whites and blacks: the Atherosclerosis Risk in Communities Study. Am. J. Epidemiol. 160, 466–474 (2004).
Garfinkel, L., Boffetta, P. & Stellman, S. D. Alcohol and breast cancer: a cohort study. Prev. Med. 17, 686–693 (1988).
Gémes, K. et al. Alcohol consumption is associated with a lower incidence of acute myocardial infarction: results from a large prospective population-based study in Norway. J. Intern. Med. 279, 365–375 (2016).
Gigleux, I. et al. Moderate alcohol consumption is more cardioprotective in men with the metabolic syndrome. J. Nutr. 136, 3027–3032 (2006).
Goldberg, R. J., Burchfiel, C. M., Reed, D. M., Wergowske, G. & Chiu, D. A prospective study of the health effects of alcohol consumption in middle-aged and elderly men. The Honolulu Heart Program. Circulation 89, 651–659 (1994).
Goldberg, R. J. et al. Lifestyle and biologic factors associated with atherosclerotic disease in middle-aged men. 20-year findings from the Honolulu Heart Program. Arch. Intern. Med. 155, 686–694 (1995).
Gordon, T. & Doyle, J. T. Drinking and coronary heart disease: the Albany Study. Am. Heart J. 110, 331–334 (1985).
Gun, R. T., Pratt, N., Ryan, P., Gordon, I. & Roder, D. Tobacco and alcohol-related mortality in men: estimates from the Australian cohort of petroleum industry workers. Aust. N.Z. J. Public Health 30, 318–324 (2006).
Harriss, L. R. et al. Alcohol consumption and cardiovascular mortality accounting for possible misclassification of intake: 11-year follow-up of the Melbourne Collaborative Cohort Study. Addiction 102, 1574–1585 (2007).
Hart, C. L. & Smith, G. D. Alcohol consumption and mortality and hospital admissions in men from the Midspan collaborative cohort study. Addiction 103, 1979–1986 (2008).
Henderson, S. O. et al. Established risk factors account for most of the racial differences in cardiovascular disease mortality. PLoS ONE 2, e377 (2007).
Hippe, M. et al. Familial predisposition and susceptibility to the effect of other risk factors for myocardial infarction. J. Epidemiol. Community Health 53, 269–276 (1999).
Ikehara, S. et al. Alcohol consumption and mortality from stroke and coronary heart disease among Japanese men and women: the Japan collaborative cohort study. Stroke 39, 2936–2942 (2008).
Ikehara, S. et al. Alcohol consumption, social support, and risk of stroke and coronary heart disease among Japanese men: the JPHC Study. Alcohol. Clin. Exp. Res. 33, 1025–1032 (2009).
Iso, H. et al. Alcohol intake and the risk of cardiovascular disease in middle-aged Japanese men. Stroke 26, 767–773 (1995).
Jakovljević, B., Stojanov, V., Paunović, K., Belojević, G. & Milić, N. Alcohol consumption and mortality in Serbia: twenty-year follow-up study. Croat. Med. J. 45, 764–768 (2004).
Keil, U., Chambless, L. E., Döring, A., Filipiak, B. & Stieber, J. The relation of alcohol intake to coronary heart disease and all-cause mortality in a beer-drinking population. Epidemiology 8, 150–156 (1997).
Key, T. J. et al. Mortality in British vegetarians: results from the European Prospective Investigation into Cancer and Nutrition (EPIC-Oxford). Am. J. Clin. Nutr. 89, 1613S–1619S (2009).
Kitamura, A. et al. Alcohol intake and premature coronary heart disease in urban Japanese men. Am. J. Epidemiol. 147, 59–65 (1998).
Kivelä, S. L. et al. Alcohol consumption and mortality in aging or aged Finnish men. J. Clin. Epidemiol. 42, 61–68 (1989).
Klatsky, A. L. et al. Alcohol drinking and risk of hospitalization for heart failure with and without associated coronary artery disease. Am. J. Cardiol. 96, 346–351 (2005).
Kono, S., Ikeda, M., Tokudome, S., Nishizumi, M. & Kuratsune, M. Alcohol and mortality: a cohort study of male Japanese physicians. Int. J. Epidemiol. 15, 527–532 (1986).
Kunutsor, S. K. et al. Self-reported alcohol consumption, carbohydrate deficient transferrin and risk of cardiovascular disease: the PREVEND prospective cohort study. Clin. Chim. Acta 520, 1–7 (2021).
Kurl, S., Jae, S. Y., Voutilainen, A. & Laukkanen, J. A. The combined effect of blood pressure and C-reactive protein with the risk of mortality from coronary heart and cardiovascular diseases. Nutr. Metab. Cardiovasc. Dis. 31, 2051–2057 (2021).
Larsson, S. C., Wallin, A. & Wolk, A. Contrasting association between alcohol consumption and risk of myocardial infarction and heart failure: two prospective cohorts. Int. J. Cardiol. 231, 207–210 (2017).
Lazarus, N. B., Kaplan, G. A., Cohen, R. D. & Leu, D. J. Change in alcohol consumption and risk of death from all causes and from ischaemic heart disease. BMJ 303, 553–556 (1991).
Lee, D.-H., Folsom, A. R. & Jacobs, D. R. Dietary iron intake and Type 2 diabetes incidence in postmenopausal women: the Iowa Women’s Health Study. Diabetologia 47, 185–194 (2004).
Liao, Y., McGee, D. L., Cao, G. & Cooper, R. S. Alcohol intake and mortality: findings from the National Health Interview Surveys (1988 and 1990). Am. J. Epidemiol. 151, 651–659 (2000).
Licaj, I. et al. Alcohol consumption over time and mortality in the Swedish Women’s Lifestyle and Health cohort. BMJ Open 6, e012862 (2016).
Lindschou Hansen, J. et al. Alcohol intake and risk of acute coronary syndrome and mortality in men and women with and without hypertension. Eur. J. Epidemiol. 26, 439–447 (2011).
Makelä, P., Paljärvi, T. & Poikolainen, K. Heavy and nonheavy drinking occasions, all-cause and cardiovascular mortality and hospitalizations: a follow-up study in a population with a low consumption level. J. Stud. Alcohol 66, 722–728 (2005).
Malyutina, S. et al. Relation between heavy and binge drinking and all-cause and cardiovascular mortality in Novosibirsk, Russia: a prospective cohort study. Lancet 360, 1448–1454 (2002).
Maraldi, C. et al. Impact of inflammation on the relationship among alcohol consumption, mortality, and cardiac events: the health, aging, and body composition study. Arch. Intern. Med. 166, 1490–1497 (2006).
Marques-Vidal, P. et al. Alcohol consumption and cardiovascular disease: differential effects in France and Northern Ireland. The PRIME study. Eur. J. Cardiovasc. Prev. Rehabil. 11, 336–343 (2004).
Meisinger, C., Döring, A., Schneider, A., Löwel, H. & KORA Study Group. Serum gamma-glutamyltransferase is a predictor of incident coronary events in apparently healthy men from the general population. Atherosclerosis 189, 297–302 (2006).
Merry, A. H. H. et al. Smoking, alcohol consumption, physical activity, and family history and the risks of acute myocardial infarction and unstable angina pectoris: a prospective cohort study. BMC Cardiovasc. Disord. 11, 13 (2011).
Miller, G. J., Beckles, G. L., Maude, G. H. & Carson, D. C. Alcohol consumption: protection against coronary heart disease and risks to health. Int. J. Epidemiol. 19, 923–930 (1990).
Mukamal, K. J., Chiuve, S. E. & Rimm, E. B. Alcohol consumption and risk for coronary heart disease in men with healthy lifestyles. Arch. Intern. Med. 166, 2145–2150 (2006).
Ng, R., Sutradhar, R., Yao, Z., Wodchis, W. P. & Rosella, L. C. Smoking, drinking, diet and physical activity-modifiable lifestyle risk factors and their associations with age to first chronic disease. Int. J. Epidemiol. 49, 113–130 (2020).
Onat, A. et al. Moderate and heavy alcohol consumption among Turks: long-term impact on mortality and cardiometabolic risk. Arch. Turkish Soc. Cardiol. 37, 83–90 (2009).
Pedersen, J. Ø., Heitmann, B. L., Schnohr, P. & Grønbaek, M. The combined influence of leisure-time physical activity and weekly alcohol intake on fatal ischaemic heart disease and all-cause mortality. Eur. Heart J. 29, 204–212 (2008).
Reddiess, P. et al. Alcohol consumption and risk of cardiovascular outcomes and bleeding in patients with established atrial fibrillation. Can. Med. Assoc. J. 193, E117–E123 (2021).
Rehm, J. T., Bondy, S. J., Sempos, C. T. & Vuong, C. V. Alcohol consumption and coronary heart disease morbidity and mortality. Am. J. Epidemiol. 146, 495–501 (1997).
Renaud, S. C., Guéguen, R., Schenker, J. & d’Houtaud, A. Alcohol and mortality in middle-aged men from eastern France. Epidemiology 9, 184–188 (1998).
Ricci, C. et al. Alcohol intake in relation to non-fatal and fatal coronary heart disease and stroke: EPIC-CVD case-cohort study. BMJ 361, k934 (2018).
Rimm, E. B. et al. Prospective study of alcohol consumption and risk of coronary disease in men. Lancet 338, 464–468 (1991).
Roerecke, M. et al. Heavy drinking occasions in relation to ischaemic heart disease mortality– an 11-22 year follow-up of the 1984 and 1995 US National Alcohol Surveys. Int. J. Epidemiol. 40, 1401–1410 (2011).
Romelsjö, A., Allebeck, P., Andréasson, S. & Leifman, A. Alcohol, mortality and cardiovascular events in a 35 year follow-up of a nationwide representative cohort of 50,000 Swedish conscripts up to age 55. Alcohol Alcohol. 47, 322–327 (2012).
Rostron, B. Alcohol consumption and mortality risks in the USA. Alcohol Alcohol. 47, 334–339 (2012).
Ruidavets, J.-B. et al. Patterns of alcohol consumption and ischaemic heart disease in culturally divergent countries: the Prospective Epidemiological Study of Myocardial Infarction (PRIME). BMJ 341, c6077 (2010).
Schooling, C. M. et al. Moderate alcohol use and mortality from ischaemic heart disease: a prospective study in older Chinese people. PLoS ONE 3, e2370 (2008).
Schutte, R., Smith, L. & Wannamethee, G. Alcohol - The myth of cardiovascular protection. Clin. Nutr. 41, 348–355 (2022).
Sempos, C., Rehm, J., Crespo, C. & Trevisan, M. No protective effect of alcohol consumption on coronary heart disease (CHD) in African Americans: average volume of drinking over the life course and CHD morbidity and mortality in a U.S. national cohort. Contemp. Drug Probl. 29, 805–820 (2002).
Shaper, A. G., Wannamethee, G. & Walker, M. Alcohol and coronary heart disease: a perspective from the British Regional Heart Study. Int. J. Epidemiol. 23, 482–494 (1994).
Simons, L. A., McCallum, J., Friedlander, Y. & Simons, J. Alcohol intake and survival in the elderly: a 77 month follow-up in the Dubbo study. Aust. N.Z. J. Med. 26, 662–670 (1996).
Skov-Ettrup, L. S., Eliasen, M., Ekholm, O., Grønbæk, M. & Tolstrup, J. S. Binge drinking, drinking frequency, and risk of ischaemic heart disease: a population-based cohort study. Scand. J. Public Health 39, 880–887 (2011).
Snow, W. M., Murray, R., Ekuma, O., Tyas, S. L. & Barnes, G. E. Alcohol use and cardiovascular health outcomes: a comparison across age and gender in the Winnipeg Health and Drinking Survey Cohort. Age Ageing 38, 206–212 (2009).
Song, R. J. et al. Alcohol consumption and risk of coronary artery disease (from the Million Veteran Program). Am. J. Cardiol. 121, 1162–1168 (2018).
Streppel, M. T., Ocké, M. C., Boshuizen, H. C., Kok, F. J. & Kromhout, D. Long-term wine consumption is related to cardiovascular mortality and life expectancy independently of moderate alcohol intake: the Zutphen Study. J. Epidemiol. Community Health 63, 534–540 (2009).
Suhonen, O., Aromaa, A., Reunanen, A. & Knekt, P. Alcohol consumption and sudden coronary death in middle-aged Finnish men. Acta Med. Scand. 221, 335–341 (1987).
Thun, M. J. et al. Alcohol consumption and mortality among middle-aged and elderly U.S. adults. N. Engl. J. Med. 337, 1705–1714 (1997).
Tolstrup, J. et al. Prospective study of alcohol drinking patterns and coronary heart disease in women and men. BMJ 332, 1244–1248 (2006).
Wannamethee, G. & Shaper, A. G. Alcohol and sudden cardiac death. Br. Heart J. 68, 443–448 (1992).
Wannamethee, S. G. & Shaper, A. G. Type of alcoholic drink and risk of major coronary heart disease events and all-cause mortality. Am. J. Public Health 89, 685–690 (1999).
Wilkins, K. Moderate alcohol consumption and heart disease. Health Rep. 14, 9–24 (2002).
Yang, L. et al. Alcohol drinking and overall and cause-specific mortality in China: nationally representative prospective study of 220,000 men with 15 years of follow-up. Int. J. Epidemiol. 41, 1101–1113 (2012).
Yi, S. W., Yoo, S. H., Sull, J. W. & Ohrr, H. Association between alcohol drinking and cardiovascular disease mortality and all-cause mortality: Kangwha Cohort Study. J. Prev. Med. Public Health 37, 120–126 (2004).
Younis, J., Cooper, J. A., Miller, G. J., Humphries, S. E. & Talmud, P. J. Genetic variation in alcohol dehydrogenase 1C and the beneficial effect of alcohol intake on coronary heart disease risk in the Second Northwick Park Heart Study. Atherosclerosis 180, 225–232 (2005).
Yusuf, S. et al. Modifiable risk factors, cardiovascular disease, and mortality in 155 722 individuals from 21 high-income, middle-income, and low-income countries (PURE): a prospective cohort study. Lancet 395, 795–808 (2020).
Zhang, Y. et al. Association of drinking pattern with risk of coronary heart disease incidence in the middle-aged and older Chinese men: results from the Dongfeng-Tongji cohort. PLoS ONE 12, e0178070 (2017).
Augustin, L. S. A. et al. Alcohol consumption and acute myocardial infarction: a benefit of alcohol consumed with meals? Epidemiology 15, 767–769 (2004).
Bianchi, C., Negri, E., La Vecchia, C. & Franceschi, S. Alcohol consumption and the risk of acute myocardial infarction in women. J. Epidemiol. Community Health 47, 308–311 (1993).
Brenner, H. et al. Coronary heart disease risk reduction in a predominantly beer-drinking population. Epidemiology 12, 390–395 (2001).
Dorn, J. M. et al. Alcohol drinking pattern and non-fatal myocardial infarction in women. Addiction 102, 730–739 (2007).
Fan, A. Z., Ruan, W. J. & Chou, S. P. Re-examining the relationship between alcohol consumption and coronary heart disease with a new lens. Prev. Med. 118, 336–343 (2019).
Fumeron, F. et al. Alcohol intake modulates the effect of a polymorphism of the cholesteryl ester transfer protein gene on plasma high density lipoprotein and the risk of myocardial infarction. J. Clin. Investig. 96, 1664–1671 (1995).
Gaziano, J. M. et al. Moderate alcohol intake, increased levels of high-density lipoprotein and its subfractions, and decreased risk of myocardial infarction. N. Engl. J. Med. 329, 1829–1834 (1993).
Genchev, G. D., Georgieva, L. M., Weijenberg, M. P. & Powles, J. W. Does alcohol protect against ischaemic heart disease in Bulgaria? A case-control study of non-fatal myocardial infarction in Sofia. Cent. Eur. J. Public Health 9, 83–86 (2001).
Hammar, N., Romelsjö, A. & Alfredsson, L. Alcohol consumption, drinking pattern and acute myocardial infarction. A case referent study based on the Swedish Twin Register. J. Intern. Med. 241, 125–131 (1997).
Hines, L. M. et al. Genetic variation in alcohol dehydrogenase and the beneficial effect of moderate alcohol consumption on myocardial infarction. N. Engl. J. Med. 344, 549–555 (2001).
Ilic, M., Grujicic Sipetic, S., Ristic, B. & Ilic, I. Myocardial infarction and alcohol consumption: a case-control study. PLoS ONE 13, e0198129 (2018).
Jackson, R., Scragg, R. & Beaglehole, R. Alcohol consumption and risk of coronary heart disease. BMJ 303, 211–216 (1991).
Kabagambe, E. K., Baylin, A., Ruiz-Narvaez, E., Rimm, E. B. & Campos, H. Alcohol intake, drinking patterns, and risk of nonfatal acute myocardial infarction in Costa Rica. Am. J. Clin. Nutr. 82, 1336–1345 (2005).
Kalandidi, A. et al. A case-control study of coronary heart disease in Athens, Greece. Int. J. Epidemiol. 21, 1074–1080 (1992).
Kaufman, D. W., Rosenberg, L., Helmrich, S. P. & Shapiro, S. Alcoholic beverages and myocardial infarction in young men. Am. J. Epidemiol. 121, 548–554 (1985).
Kawanishi, M., Nakamoto, A., Konemori, G., Horiuchi, I. & Kajiyama, G. Coronary sclerosis risk factors in males with special reference to lipoproteins and apoproteins: establishing an index. Hiroshima J. Med. Sci. 39, 61–64 (1990).
Kono, S. et al. Alcohol intake and nonfatal acute myocardial infarction in Japan. Am. J. Cardiol. 68, 1011–1014 (1991).
Mehlig, K. et al. CETP TaqIB genotype modifies the association between alcohol and coronary heart disease: the INTERGENE case-control study. Alcohol 48, 695–700 (2014).
Miyake, Y. Risk factors for non-fatal acute myocardial infarction in middle-aged and older Japanese. Fukuoka Heart Study Group. Jpn. Circ. J. 64, 103–109 (2000).
Oliveira, A., Barros, H., Azevedo, A., Bastos, J. & Lopes, C. Impact of risk factors for non-fatal acute myocardial infarction. Eur. J. Epidemiol. 24, 425–432 (2009).
Oliveira, A., Barros, H. & Lopes, C. Gender heterogeneity in the association between lifestyles and non-fatal acute myocardial infarction. Public Health Nutr. 12, 1799–1806 (2009).
Romelsjö, A. et al. Abstention, alcohol use and risk of myocardial infarction in men and women taking account of social support and working conditions: the SHEEP case-control study. Addiction 98, 1453–1462 (2003).
Schröder, H. et al. Myocardial infarction and alcohol consumption: a population-based case-control study. Nutr. Metab. Cardiovasc. Dis. 17, 609–615 (2007).
Scragg, R., Stewart, A., Jackson, R. & Beaglehole, R. Alcohol and exercise in myocardial infarction and sudden coronary death in men and women. Am. J. Epidemiol. 126, 77–85 (1987).
Tavani, A., Bertuzzi, M., Gallus, S., Negri, E. & La Vecchia, C. Risk factors for non-fatal acute myocardial infarction in Italian women. Prev. Med. 39, 128–134 (2004).
Tavani, A. et al. Intake of specific flavonoids and risk of acute myocardial infarction in Italy. Public Health Nutr. 9, 369–374 (2006).
Zhou, X., Li, C., Xu, W., Hong, X. & Chen, J. Relation of alcohol consumption to angiographically proved coronary artery disease in chinese men. Am. J. Cardiol. 106, 1101–1103 (2010).
Yang, Y. et al. Alcohol consumption and risk of coronary artery disease: a dose-response meta-analysis of prospective studies. Nutrition 32, 637–644 (2016).
Zheng, J. et al. Recent developments in Mendelian randomization studies. Curr. Epidemiol. Rep. 4, 330–345 (2017).
Mukamal, K. J., Stampfer, M. J. & Rimm, E. B. Genetic instrumental variable analysis: time to call Mendelian randomization what it is. The example of alcohol and cardiovascular disease. Eur. J. Epidemiol. 35, 93–97 (2020).
Verbanck, M., Chen, C.-Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from Mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Shi, J. et al. Mendelian randomization with repeated measures of a time-varying exposure: an application of structural mean models. Epidemiology 33, 84–94 (2022).
Burgess, S., Swanson, S. A. & Labrecque, J. A. Are Mendelian randomization investigations immune from bias due to reverse causation? Eur. J. Epidemiol. 36, 253–257 (2021).
Davey Smith, G., Holmes, M. V., Davies, N. M. & Ebrahim, S. Mendel’s laws, Mendelian randomization and causal inference in observational data: substantive and nomenclatural issues. Eur. J. Epidemiol. 35, 99–111 (2020).
Burgess, S. Violation of the constant genetic effect assumption can result in biased estimates for non-linear mendelian randomization. Hum. Hered. 88, 79–90 (2023).
Tian, H., Mason, A. M., Liu, C. & Burgess, S. Relaxing parametric assumptions for non-linear Mendelian randomization using a doubly-ranked stratification method. PLoS Genet. 19, e1010823 (2023).
Levin, M. G. & Burgess, S. Mendelian randomization as a tool for cardiovascular research: a review. JAMA Cardiol. 9, 79–89 (2024).
Burgess, S. et al. Guidelines for performing Mendelian randomization investigations: update for summer 2023. Wellcome Open Res. 4, 186 (2019).
Bowden, J., Davey Smith, G., Haycock, P. C. & Burgess, S. Consistent estimation in Mendelian randomization with some invalid instruments using a weighted median estimator. Genet. Epidemiol. 40, 304–314 (2016).
Holmes, M. V., Ala-Korpela, M. & Smith, G. D. Mendelian randomization in cardiometabolic disease: challenges in evaluating causality. Nat. Rev. Cardiol. 14, 577–590 (2017).
Labrecque, J. A. & Swanson, S. A. Interpretation and potential biases of Mendelian randomization estimates with time-varying exposures. Am. J. Epidemiol. 188, 231–238 (2019).
Spiegelman, D. et al. The Moderate Alcohol and Cardiovascular Health Trial (MACH15): design and methods for a randomized trial of moderate alcohol consumption and cardiometabolic risk. Eur. J. Prev. Cardiol. 27, 1967–1982 (2020).
DeJong, W. The Moderate Alcohol and Cardiovascular Health Trial: public health advocates should support good science, not undermine it. Eur. J. Prev. Cardiol. 28, e22–e24 (2021).
National Institutes of Health. NIH to End Funding for Moderate Alcohol and Cardiovascular Health Trial https://www.nih.gov/news-events/news-releases/nih-end-funding-moderate-alcohol-cardiovascular-health-trial (2018).
Miller, L. M., Anderson, C. A. M. & Ix, J. H. Editorial: from MACH15 to MACH0 – a missed opportunity to understand the health effects of moderate alcohol intake. Eur. J. Prev. Cardiol. 28, e23–e24 (2021).
Anderson, B. O. et al. Health and cancer risks associated with low levels of alcohol consumption. Lancet Public Health 8, e6–e7 (2023).
Au Yeung, S. L. & Lam, T. H. Unite for a framework convention for alcohol control. Lancet 393, 1778–1779 (2019).
Hernán, M. A. & Robins, J. M. Using big data to emulate a target trial when a randomized trial is not available. Am. J. Epidemiol. 183, 758–764 (2016).
Sudlow, C. et al. UK biobank: an open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 12, e1001779 (2015).
The ARIC Investigators. The Atherosclerosis Risk in Communit (ARIC) study: design and objectives. Am. J. Epidemiol. 129, 687–702 (1989).
Mahmood, S. S., Levy, D., Vasan, R. S. & Wang, T. J. The Framingham Heart Study and the epidemiology of cardiovascular disease: a historical perspective. Lancet 383, 999–1008 (2014).
Gmel, G. & Rehm, J. Measuring alcohol consumption. Contemp. Drug Probl. 31, 467–540 (2004).
Guyatt, G. H. et al. GRADE: an emerging consensus on rating quality of evidence and strength of recommendations. BMJ 336, 924–926 (2008).
Tibshirani, R. Regression shrinkage and selection via the lasso. J. R. Stat. Soc. Ser. B 58, 267–288 (1996).
Biggerstaff, B. J. & Tweedie, R. L. Incorporating variability in estimates of heterogeneity in the random effects model in meta‐analysis. Stat. Med. 16, 753–768 (1997).
Acknowledgements
Research reported in this publication was supported by the Bill & Melinda Gates Foundation [OPP1152504]. S.L. has received grants or contracts from the UK Medical Research Council [MR/T017708/1], CDC Foundation [project number 996], World Health Organization [APW No 2021/1194512], and is affiliated with the NIHR Oxford Biomedical Research Centre. The University of Oxford’s Clinical Trial Service Unit and Epidemiological Studies Unit (CTSU) is supported by core grants from the Medical Research Council [Clinical Trial Service Unit A310] and the British Heart Foundation [CH/1996001/9454]. The CTSU receives research grants from industry that are governed by University of Oxford contracts that protect its independence and has a staff policy of not taking personal payments from industry. The content is solely the responsibility of the authors and does not necessarily represent the official views of the funders. The funders of the study had no role in study design, data collection, data analysis, data interpretation, writing of the final report, or the decision to publish.
Author information
Authors and Affiliations
Contributions
S.C., S.A.M., S.I.H., and E.C.M. managed the estimation or publications process. S.C. wrote the first draft of the manuscript. S.C. had primary responsibility for applying analytical methods to produce estimates. S.C. and H.R.L. had primary responsibility for seeking, cataloging, extracting, or cleaning data; designing or coding figures and tables. S.C., D.B., S.B., E.C.M., S.I.N., J.R., and R.J.D.S. provided data or critical feedback on data sources. S.C., D.B., P.Z., A.Y.A., S.I.N., and R.J.D.S. developed methods or computational machinery. S.C., D.B., P.Z., S.B., S.I.H., E.C.M., C.J.L.M., S.I.N., J.R., R.J.D.S., S.L., and E.G. provided critical feedback on methods or results. S.C., D.B., S.A.M., S.B., S.I.H., C.J.L.M., J.R., G.A.R., S.L., and E.G. drafted the work or revised it critically for important intellectual content. S.C., S.I.H., E.C.M., and E.G. managed the overall research enterprise.
Corresponding author
Ethics declarations
Competing interests
G.A.R. has received support for this manuscript from the Bill and Melinda Gates Foundation [OPP1152504]. S.L. has received grants or contracts from the UK Medical Research Council [MR/T017708/1], CDC Foundation [project number 996], World Health Organization [APW No 2021/1194512], and is affiliated with the NIHR Oxford Biomedical Research Centre. The University of Oxford’s Clinical Trial Service Unit and Epidemiological Studies Unit (CTSU) is supported by core grants from the Medical Research Council [Clinical Trial Service Unit A310] and the British Heart Foundation [CH/1996001/9454]. The CTSU receives research grants from industry that are governed by University of Oxford contracts that protect its independence and has a staff policy of not taking personal payments from industry. All other authors declare no competing interests.
Peer review
Peer review information
Nature Communications thanks Shiu Lun Au Yeung, and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. A peer review file is available.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Source data
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article’s Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article’s Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Carr, S., Bryazka, D., McLaughlin, S.A. et al. A burden of proof study on alcohol consumption and ischemic heart disease. Nat Commun 15, 4082 (2024). https://doi.org/10.1038/s41467-024-47632-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41467-024-47632-7
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
