
Abstract
Chemical modification of proteins is an important tool for probing natural systems, creating therapeutic conjugates and generating novel protein constructs. Site-selective reactions require exquisite control over both chemo- and regioselectivity, under ambient, aqueous conditions. There are now various methods for achieving selective modification of both natural and unnatural amino acids—each with merits and limitations—providing a ‘toolkit’ that until 20 years ago was largely limited to reactions at nucleophilic cysteine and lysine residues. If applied in a biologically benign manner, this chemistry could form the basis of true Synthetic Biology.
Similar content being viewed by others


Introduction
Modification of proteins is widespread throughout nature, increasing the diversity of protein structure and hence function by up to two orders of magnitude1. Yet, our ability to synthetically mimic nature’s capacity to install such modifications is essentially limited by the chemistry that is available. Reaction at a single amino acid or site, among a sea of reactive carboxylic acids, amides, amines, alcohols and thiols, is a significant and exciting challenge in both chemo- and regioselectivity. Potential transformations, if they are to be relevant, are moulded by the need for biologically ambient conditions (that is, <37 °C, pH 6–8, aqueous solvent) so as not to disrupt protein architecture and/or function. Ideally, this should proceed with near total conversion to generate homogenous constructs2,3,4. The applications of modified proteins are many; they are as varied as the in vivo tracking of protein–fluorophore conjugates5 to the polyethylene glycol (PEG)ylation of therapeutic proteins to reduce immunogenicity6, from the production of materials with novel properties7 to probing the mechanism of pathological enzymes8.
While many past examples of so-called ‘bioconjugation’ exist (and even dedicated journals), those that teach a strong strategic lesson are more rare. The rigour of the chemical approach (including proper characterization) has been lacking—supplanted perhaps by a pragmatic desire for useful product. In an era now hungry for precise molecular knowledge of protein function, previously rare (historical) examples of precise protein chemistry become vital.
We (subjectively) consider that a seminal example can be found in the work of Wilchek9 and later Bender10 and Koshland11. Their chemical conversions of serine to cysteine have, as singularly early examples of site-directed protein mutagenesis, we believe, still not been fully appreciated. It set the stage for approaches that are only now coming to fruition in a broadly applied manner. In a post-genomic era that is more conversant with limitations of ‘gene-only’ methods, this will likely prove uniquely powerful12.
Over the past two decades, a number of methodologies have emerged (for example, Fig. 1) for undertaking modification at both natural and non-standard amino-acid residues, in vitro and in vivo, building on a previously limited toolkit for modification primarily at cysteine and lysine13. In this review, we will focus on these key developments that have greatly expanded the protein chemistry reaction palette. In particular, we will highlight merits and current limitations of reactions, in the context of the protein-conjugate bond-type formed.

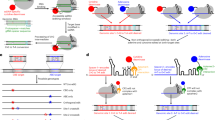
A uniquely reactive amino-acid ‘tag’171 is installed on a protein surface for reaction with a desired ‘modification’. Examples include (a) fluorophores5,125 (b) glycosylation35 (c) prenylation37 (d) PEGylation87 (e) attachment to solid surfaces172 (f) peptides44 (g) biotinylation90 and (h) antibody conjugation159.
Modifications of natural amino acids
Natural amino acids bring with them immediate accessibility without the need for more specialist techniques. Yet, the palette of functional groups is more limited and abundance may play a critical role in determining selectivity and hence precision. Low abundance (for example, cysteine) may allow (re)positioning to allow use of site-selective strategies; this is effective reassignment of the associated codons to encode the site of reaction for a complete and a hence precise alteration. Yet with high abundance, full reaction of all may be unlikely and hence reactions of all may generate (often statistical) mixtures of many products. Moreover, many of the functional groups in proteins are nucleophiles. This creates both a limitation on their use alone (differentiation based on selectivity may be more difficult) and an opportunity (unnatural amino acids (UAAs), see below, may then be designed with very different properties that are chemically distinguishable from this natural nucleophilic set).
Cysteine Sprotein–C bonds
As the most robustly nucleophilic of the 20 canonical amino acids, the thiol of cysteine offers a unique reactive handle within proteins, a property exploited extensively in nature1. Although pH may need to be controlled, selective reaction at cysteine over other nucleophilic residues such as lysine and histidine can be achieved14, while the low abundance (<2%) of cysteine in proteins often allows for facile modification at a single site3. In addition to functionalization of a protein of interest, this can also allow ready mutational repositioning and codon reassignment (through Cys→Ser/Gly mutation)15.
The selective reactions of cysteine with electrophiles such as α-halocarbonyls and maleimides have been suggested for almost a century (Fig. 2b,c)16. Indeed, iodoacetamide is used routinely for capping before digestion for protein sequencing17. Notably, some derivatives react with N-nucleophiles18. However, commercial availability and ease of use and synthesis of maleimide derivatives13,19 have led to widespread use (for example, vaccine candidates20 or modified enzymes21)13. Their use results in a reaction typically considered irreversible, yet it has been suggested that this can be reversed by competitive thiols22. Hydrolysis of the maleimide adduct moiety can also lead to subsequent decomposition of protein conjugates22,23; interestingly, this may advantageously reduce cited reversibility22. Thus, potential degradation may be an important consideration, particularly when instability may give rise to an unwanted mixture of products. Interestingly, use of bromomaleimides has opened up the possibility of reversible conjugation, allowing modulation of activity and in vivo monitoring, while also allowing the bridging and stabilisation of native disulfides19,24. The rare 21st amino acid, selenocysteine, can also be engineered into proteins and used to react with maleimides; greater Se nucleophilicity can allow conjugation selectivity over cysteine residues25.

Recently, aminoethylating agents have been used; the resulting thioether products (‘thia-lysines’) can mimic lysines bearing post-translational modifications (PTMs) such as methylation and acetylation, modifications with key roles in eukaryotic cells, particularly in the histone proteins that package DNA (Fig. 2a)26,27. More recently, transition metal-catalysed modifications of cysteine have been reported, such as rhodium-catalysed reaction with diazo compounds, although potential side reactions with tryptophan have been noted as a limitation28. Thiyl radicals at Cys can also be utilized for unique chemistries (see Box 1).
The use of cysteine nucleophiles has traditionally been limited to in vitro application due to the concentration of free thiols in cells (for example, glutathione). Some attempts have been made to conduct selective cellular labelling through introduction of Cys at particularly reactive sites of cell-surface proteins29.
Cysteine Sprotein–S bonds
The ability of sulfur to alter oxidation state is often exploited in natural redox reactions. Sulfur–sulfur bonds are key in maintaining protein tertiary and quarternary structure via interchain bridges. This property of sulfur can be exploited in the synthetic modification of proteins; formation of disulfides between thiol and cysteine can occur under an ambient atmosphere (Fig. 2e). However, rate of reaction is often slow, and disulfide exchange (ideally with kinetic control) is a favoured method for installing modifications; Ellman’s reagent has long been used to quantify free thiols.
Additional thermodynamic driving force can be exploited to favour formation of disulfide on-protein but can be unpredictable. Thus, reagents that allow kinetic control have been developed, typically relying upon thiol-specific electrophilicity. The use of methanethiosulfonates and phenylthiosulfonates has allowed quantitative, selective modification of proteins, giving labelled proteins30, modified enzymes31 and glycoconjugates32,33,34. van Kasteren et al.35 utilized such reagents to generate a mimic of P-selectin glycoprotein ligand-1, via a dual modification with a methanethiosulfonate (to generate a sulfotyrosine mimic) and triazole-forming reaction (to install a mimic of sugar sialyl-Lewisx). Selenenyl–sulfides show enhanced reactivity (Fig. 2e); S–Se intermediates can either be installed on-protein, allowing reaction with the desired thiol (electrophilic strategy), or via addition of a selenenyl–sulfide reagent to cysteine directly (nucleophilic strategy)36,37. Mechanistically, both routes appear to exploit electrophilic sulfur in the S–Se bond as a source of their chemoselectivity.
Lysine and N-terminus Nprotein–C bonds
Despite high natural abundance, lysine remains a popular choice for modification due to the number of successful reactions that can be applied to highly nucleophilic primary amines4. This is particularly the case where selectivity (as opposed to reactivity) for the site of modification is perceived not to be important, or where multiple conjugations are desired, for example, in the display of multiple antigens for creation of conjugates as putative vaccines38. Preferential conjugation with amines, over the nucleophilic thiol of cysteine, can, in principle, be achieved through use of ‘harder’ electrophiles such as activated esters39, sulfonyl chlorides13 or isothiocyanates40. Indeed, Edman degradation, the classical reaction of N-terminal protein sequencing, relies on N-terminal modification with phenyl-isothiocyanates. Unsaturated aldehyde esters are also finding increasing favour due to their ability to undergo selective irreversible azo-electrocyclizations, such as in the installation of positron-emitting-metal-binding ligands41.
An alternative, well-established reaction for modification at lysine residues is through reductive alkylation using aldehydes in the presence of sodium cyanoborohydride42. The higher stability of this reagent over sodium borohydride allows selective reaction at an appropriate pH, although the rates of reaction may be sluggish due to slow imine/iminium formation in water. Iridium catalysis utilising formate as reductant can accelerate the reaction43.
While lysine is typically the most nucleophilic amine in proteins, the N-terminus may display unique reactivity. N-terminal modification in the seminal coupling of a N-terminal cysteine and a C-terminal thioester via the ‘native chemical ligation’ (NCL) reaction is one of the most important methods for the synthetic construction of the backbone of polypeptides. Since its introduction by Kent8,44, NCL has been used extensively for the generation of synthetic proteins45. The intermolecular formation of an intermediate thioester is followed by a rapid S→N-acyl shift46, resulting in the formation of native peptide bond. Applications in fully synthetic proteins have been reviewed elsewhere45,47 and many amino acids can now be generated at the ligation site in place of cysteine48. NCL may also be utilized in the ‘semi-synthesis’ or ‘expressed protein ligation’ of proteins through the site-selective ligation of recombinantly derived thioesters and synthetic peptides49. In these latter methods thioesters are usually generated by exploiting the protein self-splicing activity of inteins50,51. Recent work by Vila-Perelló et al.52 has greatly improved the generation and purification of recombinant thioesters, allowing the semi-synthesis of proteins to gain increasing utility as a method for protein modification. The NCL reaction is perhaps illustrative of the features of success in protein chemistry since it relies in essence on enhanced, synergistic chemoselectivity derived from more than one functional group in combination (proximal amine and thiol, see Box 2). The reaction of an N-terminal Cys with cyanobenzothiazole, which ‘borrows’ from the chemoselectivity in the last step of luciferin formation, can be viewed similarly53. The N-terminus has also been used to generate uniquely reactive ketones via biomimetic transamination mediated by the co-factor pyridoxal-5′-phosphate (PLP; see section entitled Manipulating carbonyls to Cprotein=N bonds). The different pKa of the N-terminus can also be used to exploit pH-dependent chemistry with resulting differences in reactivity and selectivity.
Modifications at UAAs
UAAs present an immediately obvious opportunity to provide potentially unique chemical handles at which to undertake site-selective modification of proteins in a more broad and free-ranging way than at natural. Yet, modification of such residues can be limited by methods for their installation and the chemistry available for reaction. Here we try to give illustrative examples of both and aim to do so in a representative manner. Yet, it should be noted that many hybrid strategies that ultimately exploit the same functional groups and bond-forming processes can be considered in other ways, which are not exhaustively discussed here, such as through the use, for example, of chemoenzymatic methods to first perform an enzymatic attachment of a functional group or ‘tag’ that is then reacted.
Staudinger amide Nprotein–C bonds
The Staudinger ligation between an azide and triarylphosphine, an extension of the Staudinger reaction54, was a conceptually exciting development. Here, elegantly, an electrophilic trap was used to divert hydrolysis of the intermediate aza–ylid, generating instead a stable amide bond (Fig. 3a)55. This has allowed the modification of azido-glycoproteins incorporated into the cell-surface glycocalyx of eukaryotic cells via the sialic acid biosynthetic and metabolic machinery. Indeed, the modification of sugars in glycoproteins was subsequently shown to be applicable to the remodelling of cell surfaces in living animals, demonstrating an apparently good level of biocompatability5. With the development of techniques for site-specifically incorporating azido-amino acids, this ligation became applicable to the direct modification of protein side chains, first at auxotroph-incorporated azidohomoalanine56 and subsequently at 4-azidophenylalanine incorporated by amber-stop codon suppression57 (see Box 3). Applications of Staudinger ligation in bioconjugation have been reviewed58, with uses as diverse as fluorogenic labelling59, epitope tagging of G-protein-coupled receptors60 and the installation of photoswitches61.

Soon after the initial report, a ‘traceless’ variant was reported by the groups of Raines62 and Bertozzi63. Thus, an amide bond can be generated without residual phosphine oxide (Fig. 3b). Other variants such as a three-component Staudinger ligation have allowed the site-specific installation of amide-bonded glycomimetics64,65. More recently, Serwa et al.66 reported a phosphite-Staudinger reaction with a stalled intermediate phosphoramidate. In addition to negating the problem of possible phosphine oxidation67, this ‘traceless’ reaction valuably allows installation of phosphate mimics66.
Despite its obvious strengths, the use of the Staudinger ligation has diminished recently due to slower associated kinetics, the retained triarylphosphine oxide appendage in ‘non-traceless’ variants and problems related to phosphine oxidation and possible side reactions of phosphines in ‘traceless’ variations65,67.

Heterocycles from formal or concerted cycloadditions
In 2002, the groups of Sharpless68 and Meldal69 independently reported a stepwise modification of a classical reaction of organic chemistry, the Huisgen70–Dimroth71–Michael72 1,3-dipolar-cycloaddition between an azide and alkyne. They found that triazole formation was dramatically accelerated by the use of copper(I) even at room temperature, and was highly tolerant of both water and oxygen (Fig. 4a). The reaction was rapidly embraced by those seeking a more modular and less bespoke approach to molecular additions and has since found widespread use in the pharmaceutical and material industries, but its impact on bioconjugation has been particularly telling, initiating a number of formal and actual cycloadditions that have expanded the ability to label biological systems73.

(a) CuAAC68 and SPAAC90, and (b) IEDDA101 and ‘photo-click’112 reactions for site-selective protein modification with reported rates for small-molecule models (‘on-protein’ rates are given in brackets where reported). No rate is given for CuAAC due to the additional dependence on copper concentration.
The absence and reasonable inertness of azides and alkynes in biology has made the copper-catalysed azide–alkyne ‘cycloaddition’ (CuAAC) an excellent candidate for undertaking modification of biomolecules (although it should be noted that the CuAAC is not a true cycloaddition, rather proceeding via a metallocyclic intermediate74). In early demonstrations, Wang et al.75 coated cowpea mosaic virus with azide or alkyne moieties via nonspecific lysine labelling and then undertook CuAAC reactions to fluorescently label the capsid. Despite some limitations, such as need for organic co-solvent, unspecific labelling, incomplete conversions and breakdown in capsid structure, the selectivity of triazole formation gave a major indication of the potential power of the reaction. Soon after Speers et al.76 reported that CuAAC could be undertaken highly selectively in cellular lysates for activity-based profiling of intracellularly labelled proteins, thereby demonstrating potential tolerance towards cellular components. As with the Staudinger ligation, the ability to site-specifically incorporate UAAs into proteins allowed expanded application of the CuAAC. Having previously reported the incorporation of azide- and alkyne-containing UAAs into proteins in Escherichia coli via amber-stop codon suppression, the group of Schultz77 reported the incorporation of both O-propargyltyrosine and p-azidophenylalanine into proteins in Saccharomyces cerevisiae. They went on to demonstrate selective labelling of these UAAs using CuSO4 and copper wire (as indirect sources of Cu(I)) at 37 °C. Despite only partial conversions, these examples represented the first site-specific CuAAC on protein surfaces.
In a series of papers, the group of Tirrell78,79,80 reported that azide-containing amino acids could act as substrate surrogates for E. coli methionyl transfer RNA (tRNA) synthetase, resulting in their incorporation into cellular proteins. It was demonstrated that these ‘tagged’ proteins could undergo CuAAC to label the cell surface of E. coli for the first time78,79, as well as be used to identify newly synthesized proteins from stable cell line lysates80. This work importantly identified copper(I) bromide as a more efficient source of Cu(I) negating the need for an added reductant, yet a cellular toxicity of the metal catalyst to E. coli was suggested, with some exposed cells showing an unusual phenotype78,81. Although not yet precisely delineated, this toxicity has been attributed to the generation of reactive oxygen species that may cause intracellular damage82, implying that control of the Cu(I) oxidation state may prove key to effective reactions.
Perhaps at odds with a notion of general toxicity is the fact that several essential proteins in organisms utilize copper, leading in some cases to a relatively high cellular content83. For example, in yeast, estimates of over 105 atoms per cell have been made with a relatively low associated toxicity as judged by MIC50 of >0.7 mM84. Lack of toxicity is attributed to a highly conserved biological system for maintaining Cu(I) bound to a series of carriers, preventing the release of free copper ions that could generate reactive oxygen species. Therefore, ligands such as THPTA83, BTTES85 and histidine82 that have been used to generate Cu(I) complexes and that maintain and stabilize the metal oxidation state while also negating the potential toxicity of exogenous reductants seem a logical approach. Indeed, these have allowed the labelling of living systems including the labelling of glycans in developing zebrafish embryos85. Moreover, an alternative approach has recently been reported by Uttamapinant et al. Rather than reducing the toxicity of the catalyst, they used chelating azides, leading to a significant reduction in the required metal loading; this too allowed cell-compatible labelling86. Despite these developments, the use of CuAAC for intracellular modification in live cells is still to be reported and it is worth considering that eukaryotic cells are likely to offer additional unique challenges, which may be highly dependent on cell type.
Despite possible cellular limitations, CuAAC remains invaluable for in vitro protein modification, due to its high specificity, reasonably fast reaction rate and ease-of-use. This has led to a wide range of CuAAC reagents now being commercially available for bioconjugation. Indeed, the CuAAC can be performed site-selectively with complete conversion34 and has been used in many significant applications, such as the generation of PEGylated proteins87, the generation of dual PTM glycoprotein mimics due to its orthogonality to existing cysteine chemistry34,35, cellular proteomic analysis (BONCAT)80, a quantitative method for primary cell proteomics (QuaNCAT)88, and the construction of highly-valent protein nanoparticles89. Despite this compatibility, the perceived toxicity of copper has led to the exploration of alternative cycloaddition-type reactions.
The group of Bertozzi90 has removed copper from the equation entirely, first reporting a strain-promoted azide–alkyne cycloaddition (SPAAC) in 2004. Building on work by Wittig and Krebs91 in the 1960s, they found that highly strained cyclooctynes reacted rapidly at room temperature with azide-‘tagged’ glycoproteins in a reaction requiring no exogenous ligands or catalysts. No toxicity was observed during the reaction on the surface of mammalian cells. In its original format, the SPAAC reaction displayed similar, relatively slow, kinetics to the Staudinger ligation (Fig. 4a)67. To improve the rate, both difluorinated cyclooctynes92 (DIFO) and dibenzocylooctynes93 were independently reported allowing the visualisation of dynamic processes. In a particularly striking example, Laughlin et al.94 utilized DIFOs to visualize the development of glycans during zebrafish embryo growth, demonstrating a high degree of specificity and ‘bio-orthogonality’ at slightly faster rates than previously reported using the Staudinger ligation. Further enhancements in rates have been reported through the generation of biarylazacyclooctynones95 and cyclopropyl-fused bicyclononynes96 (Fig. 4a). The site-specific incorporation of cyclooctynes97 and biscyclononynes98 into proteins by amber-stop codon suppression has also recently been reported. However, limitations remain, such as occasional difficulty in the synthesis and handling of strained and unstable compounds, while crucially a degree of incompatibility towards cysteine has been reported99,100. In addition, these reactions remain relatively slow (Fig. 4a). As a general comment, many reported rate constants used to compare protein reactions have typically been calculated under conditions that often vary significantly, and most in fact not even on proteins but on small-molecule models; thus, direct comparison of rates should be undertaken with some caution and different derivatives may be more applicable to certain situations than others.
Inspired by the development of SPAAC reactions, the groups of Fox101 and Hilderbrand102 began to investigate the use of inverse-electron demand Diels-Alder (IEDDA) reactions as a method for bioconjugation. It was found that the reactive dienes trans-cyclooctene101 and norbornene102 react relatively rapidly with suitable tetrazine dienophiles (which release nitrogen irreversibly on subsequent retro-[4+2]-cycloaddition) allowing protein labelling at rates up to 1,000 times faster than SPAAC in the case of trans-cyclooctene (Fig. 4b). Inspired by the work of Dommerholt et al.96, it was found that trans-bicyclononene reacted at yet faster rates (while noting the caveats on rate determinations given above)103. In addition to allowing labelling of highly dynamic processes, such rapid and efficient reactions allow the concentrations of reactive partners to be lowered significantly, reducing background labelling particularly in cases where it is implausible to wash away excess reagent, such as intracellularly or in animal models104.
To generally enable IEDDA protein reactions, key reactive UAAs (tetrazine105, norbornene106,107,108, cyclooctene98,107 and biscyclononene98) have been incorporated into proteins by amber-stop codon suppression (see Box 2). Such strain-promoted cycloadditions offer intriguing and exciting possibilities for future protein labelling where the speed of labelling is vital, and recent developments suggest that the cyclooctyne-azide and cyclooctene-tetrazine reactions may have a degree of mutual compatibility, thereby allowing multi-site labelling109,110. Some limitations remain, such as the isomerisation of trans-cyclooctenes in the presence of thiols98 (cf reaction of thiols with cyclooctynes noted above) and the potential instability of tetrazines110. Also, in many variants of these SPAAC and IEDDA, reaction mixtures of regioisomers are formed (unlike the CuAAC, which is highly 1,4-selective) and in some cases bulky linkages may prevent effective syntheses of functional structures (as opposed to those that have simply been labelled) such as PTM mimics, which can often be quite small and structurally subtle.
An intriguing alternative approach to cycloadditions on proteins has been reported in a series of papers by Lin. Some tetrazoles can act as latent sources of nitrile imines, which can undergo [3+2]-cycloadditions with unactivated alkenes (Fig. 4b)111. Their generation requires irradiation with ultraviolet light (this is termed a ‘photo-click’). The reaction has been used to modify a number of alkenyl-UAAs site specifically, such as homoallylglycine112 and cyclopropenes113, while the genetic incorporation of tetrazoles has also been achieved as a reactive handle for undertaking ‘photo-click’ reactions114. While the rates of reaction are now approaching levels seen for norbornene–tetrazine conjugations, the reaction is still somewhat slower than cyclooctene–tetrazine reactions. Nonetheless, the ability to spatially and temporally control the reaction through light makes the ‘photo-click’ an attractive alternative for the site-specific labelling of proteins113.
Metal-mediated Cprotein–C or Cprotein=C bonds
Transition metal (TM) catalysis has revolutionized organic synthesis with the ability to tune reactivity by careful choice of metal, ligand and reaction conditions, allowing the generation of previously inaccessible carbon and heteroatom-containing scaffolds. Many of the factors that make such reactions appealing to the synthetic chemist also make them attractive for protein modification115. Such reactions are often associated with excellent functional group tolerance and high yields under mild conditions, while the reactive handles utilized are often inert in biological systems. However, restrictions such as a need to proceed efficiently at low protein loadings, solely in aqueous media, and to tolerate potential nonspecific binding to the multitude of possible Lewis-basic residues on protein surfaces, until recently, hindered the use of such reactions for site-specific protein modification.
Arguably, the most widely used TM-catalysed reactions in organic synthesis are the series of palladium-catalysed sp2–sp2 coupling reactions between aryl/alkenyl halides and a variety of coupling partners such as boronic acids (Suzuki–Miyaura), alkenes (Mizoroki–Heck) and alkynes (Sonogashira). Early examples of couplings on short synthetic peptides required high temperatures, or the presence of organic solvents, yet demonstrated tolerance of Pd towards some amino-acid functional groups116,117. Despite the UAA p-iodophenylalanine having been incorporated into proteins by amber-stop codon suppression and proposed as a Pd coupling partner as early as 2002 (ref. 118), it was not until 2006 that this was partially realized by Kodama et al. Both Heck and Sonagashira reactions were demonstrated, albeit in low yields (2% Heck, 25% Sonagashira), representing early examples of Pd-catalysed couplings on polypeptidic substrates119,120. Brustad et al.121 then went on to demonstrate that p-boronophenylalanine could be used to undertake Suzuki couplings, although again low yields (30%) and high temperatures (70 °C) that caused protein denaturation limited the usefulness of the reaction. It was not until 2009 that Chalker et al.122 demonstrated the first efficient Pd-mediated reaction on a protein, through the discovery of a water-and-air-stable ligand, 2-amino-4,6-dihydroxypyrimidine (ADHP, L1), for undertaking Suzuki–Miyaura cross-couplings at 37 °C in water at pH 8 (Fig. 5a). This allowed a variety of boronic acids to be coupled to a model cysteine-linked aryl iodide, with the benefit that even hydrophobic moieties could be transferred to the protein surface due to the water-solubilizing effect of the boronate group. To generalize this reaction to genetically incorporated amino acids, Spicer and Davis123 (and later Liu et al.124) demonstrated that through amber-stop codon suppression, p-iodophenylalanine could be used as a reactive handle for protein Suzuki–Miyaura cross-coupling. During this work, previously hypothesized weak, nonspecific binding of TMs to Lewis-basic amino acids was encountered under some conditions, leading to ambiguity in reaction analysis; this was circumvented by the identification of a suitable palladium scavenger. The group of Davis has since gone on to demonstrate that the Suzuki–Miyaura reaction is applicable to couplings on the cell surface of E. coli, demonstrating a negligible catalyst toxicity125, and that the coupling of carbohydrate–boronic acids to cell surfaces can be used to mimic glycoproteins in a cellular synthetic glycocalyx125,126.

Although ADHP is an efficient catalyst for undertaking Suzuki–Miyaura cross-couplings, it can be less effective for other Pd-catalysed reactions. Li et al.127 have since shown that by simple methylation of the ligand, the same catalytic system could be used to promote Sonogashira reactions (L2 in Fig. 5a), while the minimal motif guanidine-based ligands (L3 and L4 in Fig. 5a) can significantly enhance the rate of Suzuki reactions relative to ADHP128 and allow efficient couplings even at low stoichiometries and concentrations suitable when labelling with scarce reagents129. It was also shown that PEG chains offer new reactivity and a significant enhancement in rate as self-liganding (internally chelating) boronic acids when used with Pd(OAc)2 to give a high-yielding site-specific PEGylation of proteins128. This self-liganded effect has more recently been exploited by Li et al.130, who found that PEG-conjugated fluorophores with Pd(NO3)2 could efficiently catalyse the Sonogashira cross-coupling, even intracellularly, in E. coli and Shigella.
Olefin metathesis has also found recent application in the site-selective modification of proteins, due to the discovery by Lin et al.131 that allyl sulfides are privileged substrates for undertaking aqueous cross-metathesis with Hoveyda–Grubbs II catalyst, via a proposed sulfur-relayed mechanism (Fig. 5b). The subsequent use of a variety of olefinic amino-acid side chains containing allylic heteroatoms suggested a breadth for this allylic chalcogen effect132. This allowed the installation of a number of olefin substrates including PEG and allyl glycosides at an S-allyl cysteine residue, introduced into proteins via a number of chemical routes133. Determination of sensitivity to accessibility, self-metathesis and reagent reactivity has delineated predictive rules for this protein reaction134. Moreover, tuning of heteroatom (S→Se) in Se-allylselenocysteine led to a significant increase in reaction rate and expanded substrate scope135; this was applied to a chemically controlled ‘write-read-erase’ histone protein modification cycle.
A further example of intriguing TM catalysis was first reported by Antos and Francis136, utilising rhodium-generated carbenoids formed from diazo reagents for modification of tryptophan residues. While this reaction initially required quite harsh acidic conditions (pH 3), it was subsequently found that this was primarily to denature early protein substrates and hence to expose the reactive tryptophan residue; conjugations at pH 6 are now possible137. Recent reports by the group of Ball138,139 have used rhodium-bound metallopeptides to catalyse modification of tryptophan by using a structure-directed approach. Despite this elegantly designed rate enhancement, the need for a highly specific interaction to direct the reaction will likely limit its general applicability. However, it represents an impressive example of molecular recognition to override inherent reactivity.
Given the strength and ubiquity of carbon–carbon bonds, there will be continuing utility in their formation, despite the potential of heteroatom–carbon bond-forming chemistry. We have focused in this section on metal-mediated processes since, thus far, they have dominated current strategies, yet it should be noted that other possible strategies exist that exploit non-metal-mediated processes, such as aldol140 or Wittig chemistry141, the use of (formal) cycloaddition chemistry or as the result of a relay from a prior bond-forming event (for example, Pictet–Spengler).
Manipulating carbonyls to Cprotein=N bonds
Despite being widespread throughout nature, the carbonyl groups of aldehydes and ketones are almost entirely absent from native proteins142. Yet, the diversity of unique chemistry that they can undergo in the presence of the natural functional groups of proteins makes them an attractive handle for protein modification. They have found particular use in the reaction of hydrazines and hydroxylamines to form hydrazones and oximes, respectively, under acidic conditions (in part due to reagent availability and ease of use). These reactions can be accelerated by nucleophilic catalysts such as aniline143.
To install aldehydes and ketones into proteins, a number of methods have been identified. Among the earliest was the discovery that periodate oxidation (cleavage) of N-terminal Ser/Thr residues led to a terminal aldehyde, which could then react selectively with a fluorescent hydrazine to allow site-specific protein tagging144. The group of Francis145 has utilized a biomimetic PLP-mediated transamination to generate N-terminal ketones. Investigation of the reaction conditions indicates that a range of amino acids are tolerated by this reaction146, allowing the selective modification of antibodies147 and filamentous phage148.
The genetic incorporation of a ketone-containing amino acid by amber-stop codon suppression was first reported by Cornish et al.149 via chemical acylation of a tRNA synthetase. This residue, once installed, again reacted selectively with a range of fluorescent hydrazines. The ketone amino acids p-acetylphenylalanine142 and m-acetylphenylalanine150 were subsequently incorporated into proteins, without the need for chemical acylation, in both E. coli142,150 and eukaryotic cells151. More recently, Huang et al.152 reported the incorporation of aliphatic ketone-containing amino acids that showed improved reaction kinetics, while diketone-containing amino acids have been reported to give increased stability in oxime products153. Alternatively, Carrico et al.154 have exploited a six-residue sequence tag that directs a natural formylglycine-generating enzyme in both prokaryotic and eukaryotic cells.
The subsequent formation of hydrazones and oximes from these carbonyls has found widespread use in the conjugation of functional handles and probes to proteins. For example, hydroxylamines have been used to generate glycoprotein mimics155, to label G-protein-coupled receptors156, antibodies157 and therapeutic proteins for increased pharmacokinetics6, and for dual protein tagging and formation of bifunctional antibodies158,159. However, despite its widespread use, the reactions of aldehydes and ketones suffer from a number of key drawbacks. Most importantly, due to the presence of a range of carbonyl-containing substrates in cells, this chemistry is not necessarily suitable for in vivo applications. In addition, the hydrazone and oxime linkages are inherently unstable, leading to hydrolysis over the course of hours, particularly under acidic conditions, although hydroxylamines are reported to generate more stable linkages160. To avoid such instability, Sasaki et al.161 recently reported the use of a modified Pictet–Spengler reaction for aldehyde modification (which ultimately leads to the subsequent creation of C–C and C–N bonds), with Agarwal et al.162 subsequently reporting greatly improved reaction kinetics. Although the fundamental limitation of a lack of bioapplicability still remains, the facile use of aldehyde/ketone chemistry remains an attractive tool for in vitro modifications.
Cprotein–S/Se bonds
The UAA dehydroalanine (Dha) can be used as a Michael acceptor and has found extensive use in protein modification, reacting rapidly with sulfur nucleophiles to generate alkyl cysteine analogues, offering an electrophilic alternative to nucleophilic reaction of cysteine3. This is particularly useful in examples where use of electrophilic alkylation of Cys is difficult to control or where appropriate electrophiles cannot be generated, and leads to a greater level of selectivity. Dha can be accessed via a number of routes: elimination of active-site serines, the oxidative elimination of unnatural selenocysteine amino acids163,164 or through the milder oxidative elimination of cysteine with sulfonylhydroxylamine reagents165. All can prove efficient but occasional side reactions in all recently prompted the development of a bis-alkylation method: cyclic sulfoniums can be eliminated to form Dha under strikingly mild conditions166.
The addition of functionalized thiols to Dha takes place rapidly and selectively under mild conditions. This reaction has been used to install a number of thioether mimics of natural protein modifications such as lipidation, glycosylation, phosphorylation and lysine methylation/acetylation164,165,167,168, as well as installing reactive handles for further modification such as S-allyl cysteine for olefin metathesis.131 These reactions typically (but not always) proceed with low substrate control in their diastereoselection and so a mixture of D/L-epimers is produced at the site of modification. Dha also provides a viable method for chemically creating selenocysteines in proteins135. Although only shown for a single example (Se-allyl cysteine), the discovery of conditions for creating suitable Se nucleophiles for this addition may enable broader methods.
While the olefin in Dha displays unique conjugate electrophile reactivity, isolated olefins in, for example, homoallylglycine can serve as useful UAAs for modifications using radical chemistry (see Box 1)169.
Outlook and future directions
The introduction of site-directed gene mutagenesis as a powerful method for altering protein structure at a genetic level revolutionized the study and application of proteins. The ability to switch between natural amino acids at virtually any desired residue site in a recombinant protein has allowed unparalleled progress in the manipulation of proteins for scientific discovery. Yet, this ability to access and alter functionality is limited by the 20 typical proteinogenic amino acids and a limited palette of chemical functional groups. As such, there is a powerful need for chemical modification of proteins and the installation of non-natural functionality as a strategy for more free-ranging protein synthesis or design. New methods should aspire to the widespread success and applicability of gene mutagenesis as a tool in the biological sciences12.
Over the past 15 years, the field of chemical protein modification has been dramatically revitalized, from one that focused on the use of natural cysteine and lysine residues to one that now utilizes a wide range of chemical handles, coupling partners and conditions, many of which are mutually compatible (‘orthogonal’) not just with (to) each other, thereby allowing multiple modifications to be undertaken, but also with (to) living systems, allowing them to be utilized in vivo170.
The use of such reactions is only beginning to be exploited as improvements in selectivity, kinetics, compatibility and ease of use are made. The potential applications of modified proteins are virtually limitless, whether be it for the in vivo tracking of dynamic processes, the conjugation of therapeutic agents, the elucidation of biosynthetic/metabolic pathways or the use of modified protein-based materials with novel functionality and structure. The development of these reactions has been reviewed here from the viewpoint of applicable chemistry, rather than the biological uses of modified proteins, and it is likely that with an expanding toolkit of chemical reactions for installing a range of modifications, chemists and biologists will discover exciting applications as yet unexplored. This could truly become an unlimited form of Synthetic Biology.
Since such protein methods have become a ‘hot-topic’ over the past decade, it has been easy to forget some critical principles in developing chemical reactions for modifying proteins. Sadly, increasing numbers of reports are now simply undertaking reactions ‘because one can’ with little-or-no regard for improving (either strategically or functionally) on the plethora of reactions already available. We see a particular need to develop chemical reactions that allow: the selective installation of a desired functionality in a manner that allows the mimicry of a natural modification, the rapid labelling of a biologically relevant site or new in vivo reactivity. We see less value in the discovery of those reactions that may not have been performed on a protein previously but in fact offer no benefit compared with the existing ‘toolkit’. Put more succinctly, there is not necessarily a need for new chemistry for modifying proteins; there is a need for better chemistry for modifying proteins. It is important to remember that proteins should not be seen merely as a substrate for undertaking a reaction. Rather, this chemistry should be increasingly seen as a method for testing a hypothesis, developing the technology or creating a functional probe.
To this extent, a number of key challenges can be envisaged that must be addressed in future developments in the field. While the reactions discussed here represent useful discoveries that will undoubtedly make a large impact on protein science, they are not without their limitations. A reaction that combines the ‘selling points’ of each must be seen as highly desirable: the mimicry and minimal linker afforded by cysteine, the ease of modification at lysine, the bio-orthogonality of the Staudinger ligation, the unparalleled speed of cycloadditions, the tunability of transition metal-mediated reactions, the potential reversibility and switching of carbonyl chemistry. A reaction that combines these favourable characteristics may represent an ideological end goal in the development of new chemistries. When judged by these criteria of utility, protein chemistry is still in its infancy. As it matures, it will likely revolutionize the molecular analysis of Biology.
Additional information
How to cite this article: Spicer, C. D. and Davis, B. G. Selective chemical protein modification. Nat. Commun. 5:4740 doi: 10.1038/ncomms5740 (2014).
References
Walsh, C. T. Posttranslational Modification of Proteins: Expanding Natures Inventory Roberts and Company Publishers (2006).
de Graaf, A. J., Kooijman, M., Hennink, W. E. & Mastrobattista, E. Nonnatural amino acids for site-specific protein conjugation. Bioconjug. Chem. 20, 1281–1295 (2009).
Chalker, J. M., Bernardes, G. J., Ya, L. & Davis, B. G. Chemical modification of proteins at cysteine: opportunities in chemistry and biology. Chem. Asian J. 4, 630–640 (2009).
Sletten, E. M. & Bertozzi, C. R. Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. 48, 6974–6998 (2009).
Prescher, J. A., Dube, D. H. & Bertozzi, C. R. Chemical remodelling of cell surfaces in living animals. Nature 430, 873–877 (2004).The Staudinger ligation was used to undertake chemical modification of cell surfaces in mice for the first time; a powerful early example of in vivo chemistry.
Cho, H. et al. Optimized clinical performance of growth hormone with an expanded genetic code. Proc. Natl Acad. Sci. USA 108, 9060–9065 (2011).
Witus, L. S. & Francis, M. B. Using synthetically modified proteins to make new materials. Acc. Chem. Res. 44, 774–783 (2011).
Schnolzer, M. & Kent, S. B. Constructing proteins by dovetailing unprotected synthetic peptides: backbone-engineered HIV protease. Science 256, 221–225 (1992).
Zioudrou, C., Wilchek, M. & Patchornik, A. Conversion of the L-serine residue to an L-cysteine residue in peptides. Biochemistry 4, 1811–1822 (1965).We consider this paper pivotal in priming the concept of convergent protein synthesis; the ability to selectively ‘chemically mutate’/‘post-expression mutate’ one residue to another has had long standing implications.
Polgar, L. & Bender, M. L. A new enzyme containing a synthetically formed active site. Thiol-subtilisin. J. Am. Chem. Soc. 88, 3153–3154 (1966).
Neet, K. E. & Koshland, D. E. Jr The conversion of serine at the active site of subtilisin to cysteine: a “chemical mutation”. Proc. Natl Acad. Sci. USA 56, 1606–1611 (1966).
Chalker, J. M. & Davis, B. G. Chemical mutagenesis: selective post-expression interconversion of protein amino acid residues. Curr. Opin. Chem. Biol. 14, 781–789 (2010).
Hermanson, G. T. Bioconjugate Techniques 2nd edn Academic Press, Inc. (2008).
Crankshaw, M. W. & Grant, G. A. Modification of Cysteine Wiley (1996).
Clark, P. I. & Lowe, G. Chemical mutations of papain. The preparation of Ser 25- and Gly 25-Papain. J. Chem. Soc. Chem. Commun. 24, 923–924 (1977).
Goddard, D. R. & Michaelis, L. Derivatives of keratin. J. Biol. Chem. 112, 361–371 (1935).
Lundell, N. & Schreitmüller, T. Sample preparation for peptide mapping--A pharmaceutical quality-control perspective. Anal. Biochem. 266, 31–47 (1999).
Stephanopoulos, N., Tong, G. J., Hsiao, S. C. & Francis, M. B. Dual-surface modified virus capsids for targeted delivery of photodynamic agents to cancer cells. ACS Nano 4, 6014–6020 (2010).
Smith, M. E. B. et al. Protein modification, bioconjugation, and disulfide bridging using bromomaleimides. J. Am. Chem. Soc. 132, 1960–1965 (2010).
Betting, D. J., Kafi, K., Abdollahi-fard, A., Hurvitz, S. A. & Timmerman, J. M. Sulfhydryl-based tumor antigen-carrier protein conjugates stimulate superior antitumor immunity against B cell lymphomas. J. Immunol. 181, 4131–4140 (2008).
Zhang, Y., Bhatt, V. S., Sun, G., Wang, P. G. & Palmer, A. F. Site-selective glycosylation of hemoglobin on Cys beta93. Bioconjug. Chem. 19, 2221–2230 (2008).
Shen, B.-Q. et al. Conjugation site modulates the in vivo stability and therapeutic activity of antibody-drug conjugates. Nat. Biotechnol. 30, 184–189 (2012).
Nathani, R. I. et al. Reversible protein affinity-labelling using bromomaleimide-based reagents. Org. Biomol. Chem. 11, 2408–2411 (2013).
Moody, P. et al. Bromomaleimide-linked bioconjugates are cleavable in mammalian cells. Chembiochem 13, 39–41 (2012).
Hofer, T., Thomas, J. D., Terrence, R., Burke, J. & Rader, C. An engineered selenocysteine defines a unique class of antibody derivatives. Proc. Natl Acad. Sci. USA 105, 12451–12456 (2008).
Simon, M. D. et al. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell 128, 1003–1012 (2007).
Chatterjee, C. & Muir, T. W. Chemical approaches for studying histone modifications. J. Biol. Chem. 285, 11045–11050 (2010).
Kundu, R. & Ball, Z. T. Rhodium-catalyzed cysteine modification with diazo reagents. Chem. Commun. 2, 4166–4168 (2012).
Bös, C., Lorenzen, D. & Braun, V. Specific in vivo labeling of cell surface-exposed protein loops: reactive cysteines in the predicted gating loop mark a ferrichrome binding site and a ligand-induced conformational change of the Escherichia coli FhuA protein. J. Bacteriol. 180, 605–613 (1998).
Kenyon, G. L. & Bruice, T. W. Methods Enzymol. 47, 407–430 (1977).
Berglund, P. et al. Chemical modification of cysteine mutants of subtilisin bacillus lentus can create better catalysts than the wild-type enzyme. J. Am. Chem. Soc. 119, 5265–5266 (1997).A far-sighted example of using site-selective modification methods to create a logical array of homogenous and precise enzyme variants; here catalytic activity was modulated in a direct manner.
Davis, B. G., Maughan, M. A. T., Green, M. P., Ullman, A. & Jones, J. B. Glycomethanethiosulfonates: powerful reagents for protein glycosylation. Tetrahedron Asymmetry 11, 245–262 (2000).
Gamblin, D. P. et al. Glycosyl phenylthiosulfonates (glyco-PTS): novel reagents for glycoprotein synthesis. Org. Biomol. Chem. 1, 3642–3644 (2003).
van Kasteren, S. I., Kramer, H. B., Gamblin, D. P. & Davis, B. G. Site-selective glycosylation of proteins: creating synthetic glycoproteins. Nat. Protoc. 2, 3185–3194 (2007).
van Kasteren, S. I. et al. Expanding the diversity of chemical protein modification allows post-translational mimicry. Nature 446, 1105–1109 (2007).Two mutually compatible reactions were applied to create different site-selective modifications (S–S and triazole) in di-modified proteins; these acted as effective mimics of natural modifications in vitro and in vivo.
Gamblin, D. P. et al. Glyco-SeS: selenenylsulfide-mediated protein glycoconjugation--a new strategy in post-translational modification. Angew. Chem. Int. Ed. 43, 828–833 (2004).
Gamblin, D. P. et al. Chemical site-selective prenylation of proteins. Mol. Biosyst. 4, 558–561 (2008).
Smith, M. L. et al. Modified tobacco mosaic virus particles as scaffolds for display of protein antigens for vaccine applications. Virology 348, 475–488 (2006).
Kalkhof, S. & Sinz, A. Chances and pitfalls of chemical cross-linking with amine-reactive N-hydroxysuccinimide esters. Anal. Bioanal. Chem. 392, 305–312 (2008).
Nakamura, T., Kawai, Y., Kitamoto, N., Osawa, T. & Kato, Y. Covalent modification of lysine residues by allyl isothiocyanate in physiological conditions: plausible transformation of isothiocyanate from thiol to amine. Chem. Res. Toxicol. 22, 536–542 (2009).
Tanaka, K. et al. A submicrogram-scale protocol for biomolecule-based PET imaging by rapid 6pi-azaelectrocyclization: visualization of sialic acid dependent circulatory residence of glycoproteins. Angew. Chem. Int. Ed. 47, 102–105 (2008).
Jentoft, N. & Dearborn, D. G. Labeling of proteins by reductive methylation using sodium cyanoborohydride. J. Biol. Chem. 254, 4359–4365 (1979).
McFarland, J. M. & Francis, M. B. Reductive alkylation of proteins using iridium catalyzed transfer hydrogenation. J. Am. Chem. Soc. 127, 13490–13491 (2005).
Dawson, P. E., Muir, T. W., Clark-Lewis, I. & Kent, S. B. H. Synthesis of proteins by native chemical ligation. Science 266, 776–778 (1994).This paper created the benchmark for use of native amide bond formation under protein compatible conditions (native chemical ligation) to join two synthetic peptides to produce full length protein; a prime example of protein synthesis through linear assembly.
Kent, S. B. H. Total chemical synthesis of proteins. Chem. Soc. Rev. 38, 338–351 (2009).
Wieland, T., Bokelmann, E., Bauer, L., Lang, H. U. & Lau, H. Uber Peptidsynthesen. 8. Mitteilung Bildung von S-haltingen Peptiden durch intromolekulare Wanderung von Aminoacylresten. Liebigs Ann. Chem. 583, 129–149 (1953).
Nilsson, B. L., Soellner, M. B. & Raines, R. T. Chemical synthesis of proteins. Annu. Rev. Biophys. Biomol. Struct. 34, 91–118 (2005).
Dawson, P. E. Native chemical ligation combined with desulfurization and deselenization: a general strategy for chemical protein synthesis. Isr. J. Chem. 51, 862–867 (2011).
Muir, T. W., Sondhi, D. & Cole, P. A. Expressed protein ligation: a general method for protein engineering. Proc. Natl Acad. Sci. USA 95, 6705–6710 (1998).
Vila-Perelló, M. & Muir, T. W. Biological applications of protein splicing. Cell 143, 191–200 (2010).
Komarov, A. G., Linn, K. M., Devereaux, J. J. & Valiyaveetil, F. I. Modular strategy for the semisynthesis of a K+ channel: investigating interactions of the pore helix. ACS Chem. Biol. 4, 1029–1038 (2009).
Vila-Perelló, M. et al. Streamlined expressed protein ligation using split inteins. J. Am. Chem. Soc. 135, 286–292 (2013).
Ren, H. et al. A biocompatible condensation reaction for the labeling of terminal cysteine residues on proteins. Angew. Chem. Int. Ed. 48, 9658–9662 (2009).
Staudinger, H. & Meyer, J. Über neue organische Phosphorverbindungen III. Phosphinmethylenderivate und Phosphinimine. Helv. Chim. Acta 2, 635–646 (1919).
Saxon, E. & Bertozzi, C. R. Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010 (2000).
Kiick, K. L., Saxon, E., Tirrell, D. A. & Bertozzi, C. R. Incorporation of azides into recombinant proteins for chemoselective modification by the Staudinger ligation. Proc. Natl Acad. Sci. USA 99, 19–24 (2002).
Tsao, M.-L., Tian, F. & Schultz, P. G. Selective Staudinger modification of proteins containing p-azidophenylalanine. Chembiochem 6, 2147–2149 (2005).
van Berkel, S. S., van Eldijk, M. B. & van Hest, J. C. M. Staudinger ligation as a method for bioconjugation. Angew. Chem. Int. Ed. 50, 8806–8827 (2011).
Lemieux, G. a., De Graffenried, C. L. & Bertozzi, C. R. A fluorogenic dye activated by the staudinger ligation. J. Am. Chem. Soc. 125, 4708–4709 (2003).
Naganathan, S., Ye, S., Sakmar, T. P. & Huber, T. Site-specific epitope tagging of G protein-coupled receptors by bioorthogonal modification of a genetically encoded unnatural amino acid. Biochemistry 52, 1028–1036 (2013).
Szymański, W., Wu, B., Poloni, C., Janssen, D. B. & Feringa, B. L. Azobenzene photoswitches for Staudinger-Bertozzi ligation. Angew. Chem. Int. Ed. 125, 2122–2126 (2013).
Nilsson, B. L., Kiessling, L. L. & Raines, R. T. Staudinger ligation: a peptide from a thioester and azide. Org. Lett. 2, 1939–1941 (2000).
Saxon, E., Armstrong, J. I. & Bertozzi, C. R. A. “Traceless” Staudinger ligation for the chemoselective synthesis of amide bonds. Org. Lett. 2, 2141–2143 (2000).
Doores, K. J. et al. Direct deprotected glycosyl-asparagine ligation. Chem. Commun. 7, 1401–1403 (2006).
Bernardes, G. J. L., Linderoth, L., Doores, K. J., Boutureira, O. & Davis, B. G. Site-selective traceless Staudinger ligation for glycoprotein synthesis reveals scope and limitations. Chembiochem 12, 1383–1386 (2011).
Serwa, R. et al. Chemoselective Staudinger-phosphite reaction of azides for the phosphorylation of proteins. Angew. Chem. Int. Ed. 48, 8234–8239 (2009).
Agard, N. J., Baskin, J. M., Prescher, J. A., Lo, A. & Bertozzi, C. R. A comparative study of bioorthogonal reactions with azides. ACS Chem. Biol. 1, 644–648 (2006).
Rostovtsev, V. V., Green, L. G., Fokin, V. V. & Sharpless, K. B. A stepwise huisgen cycloaddition process: copper(I)-catalyzed regioselective "ligation" of azides and terminal alkynes. Angew. Chem. Int. Ed. 41, 2596–2599 (2002).
Tornøe, C. W., Christensen, C. & Meldal, M. Peptidotriazoles on solid phase: [1,2,3]-triazoles by regiospecific copper(i)-catalyzed 1,3-dipolar cycloadditions of terminal alkynes to azides. J. Org. Chem. 67, 3057–3064 (2002).
Huisgen, R. 1,3-Dipolar cycloadditions past and future. Angew. Chem. Int. Ed. 2, 566–598 (1963).
Dimroth, O. Synthesen mit Diazobenzolimid. Ber. Dtsch. Chem. Ges. 36, 909–913 (1903).
Michael, A. Ueber die Einwirkung von Diazobenzolimid auf Acetylendicarbonsäuremethylester. J. Prakt. Chem. 48, 94–95 (1893).
Meldal, M. & Tornøe, C. W. Cu-catalyzed azide-alkyne cycloaddition. Chem. Rev. 108, 2952–3015 (2008).
Himo, F. et al. Copper(I)-catalyzed synthesis of azoles. DFT study predicts unprecedented reactivity and intermediates. J. Am. Chem. Soc. 127, 210–216 (2005).
Wang, Q. et al. Bioconjugation by copper(I)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 125, 3192–3193 (2003).
Speers, A. E., Adam, G. C. & Cravatt, B. F. Activity-based protein profiling in vivo using a copper(i)-catalyzed azide-alkyne [3+2] cycloaddition. J. Am. Chem. Soc. 125, 4686–4687 (2003).
Deiters, A. et al. Adding amino acids with novel reactivity to the genetic code of Saccharomyces cerevisiae. J. Am. Chem. Soc. 125, 11782–11783 (2003).
Link, J. & Tirrell, D. Cell surface labeling of Escherichia coli via copper(I)-catalyzed [3+2] cycloaddition. J. Am. Chem. Soc. 125, 11164–11165 (2003).CuAAC used for the selective modification of proteins on cell (E. coli) surfaces.
Link, J., Vink, M. K. S. & Tirrell, D. Presentation and detection of azide functionality in bacterial cell surface proteins. J. Am. Chem. Soc. 126, 10598–10602 (2004).
Dieterich, D. C., Link, J., Graumann, J., Tirrell, D. & Schuman, E. M. Selective identification of newly synthesized proteins in mammalian cells using bioorthogonal noncanonical amino acid tagging (BONCAT). Proc. Natl Acad. Sci. USA 103, 9482–9487 (2006).
Link, J. et al. Discovery of aminoacyl-tRNA synthetase activity through cell-surface display of noncanonical amino acids. Proc. Natl Acad. Sci. USA 103, 10180–10185 (2006).
Kennedy, D. C. et al. Cellular consequences of copper complexes used to catalyze bioorthogonal click reactions. J. Am. Chem. Soc. 133, 17993–18001 (2011).
Hong, V., Steinmetz, N. F., Manchester, M. & Finn, M. G. Labeling live cells by copper-catalyzed alkyne--azide click chemistry. Bioconjug. Chem. 21, 1912–1916 (2010).
Rae, T. D., Schmidt, P. J., Pufahl, R. A., Culotta, V. C. & V. O'Halloran, T. Undetectable intracellular free copper: the requirement of a copper chaperone for superoxide dismutase. Science 284, 805–808 (1999).
Soriano Del Amo, D. et al. Biocompatible copper(I) catalysts for in vivo imaging of glycans. J. Am. Chem. Soc. 132, 16893–16899 (2010).
Uttamapinant, C. et al. Fast, cell-compatible click chemistry with copper-chelating azides for biomolecular labeling. Angew. Chem. Int. Ed. 51, 5852–5856 (2012).
Deiters, A., Cropp, T. A., Summerer, D., Mukherji, M. & Schultz, P. G. Site-specific PEGylation of proteins containing unnatural amino acids. Bioorg. Med. Chem. Lett. 14, 5743–5745 (2004).
Howden, A. J. M. et al. QuaNCAT: quantitating proteome dynamics in primary cells. Nat. Methods 10, 343–346 (2013).
Ribeiro-Viana, R. et al. Virus-like glycodendrinanoparticles displaying quasi-equivalent nested polyvalency upon glycoprotein platforms potently block viral infection. Nat. Commun. 3, 1303 (2012).
Agard, N. J., Prescher, J. a. & Bertozzi, C. R. A strain-promoted [3+2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc. 126, 15046–15047 (2004).
Wittig, G. & Krebs, A. Zur Existenz niedergliedriger cycloalkine, I. Chem. Ber. 94, 3260–3275 (1961).
Baskin, J. M. et al. Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl Acad. Sci. USA 104, 16793–16797 (2007).
Ning, X., Guo, J., Wolfert, M. A. & Boons, G.-J. Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew. Chem. Int. Ed. 47, 2253–2255 (2008).
Laughlin, S. T., Baskin, J. M., Amacher, S. L. & Bertozzi, C. R. In vivo imaging of membrane-associated glycans in developing zebrafish. Science 320, 664–667 (2008).
Jewett, J. C., Sletten, E. M. & Bertozzi, C. R. Rapid Cu-free click chemistry with readily synthesized biarylazacyclooctynones. J. Am. Chem. Soc. 132, 3688–3690 (2010).
Dommerholt, J. et al. Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew. Chem. Int. Ed. 49, 9422–9425 (2010).
Plass, T., Milles, S., Koehler, C., Schultz, C. & Lemke, E. A. Genetically encoded copper-free click chemistry. Angew. Chem. Int. Ed. 50, 3878–3881 (2011).
Lang, K. et al. Genetic encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels-Alder reactions. J. Am. Chem. Soc. 134, 10317–10320 (2012).
Chang, P. V. et al. Copper-free click chemistry in living animals. Proc. Natl Acad. Sci. USA 107, 1821–1826 (2010).
Lo Conte, M. et al. Multi-molecule reaction of serum albumin can occur through thiol-yne coupling. Chem. Commun. 47, 11086–11088 (2011).
Blackman, M. L., Royzen, M. & Fox, J. M. Tetrazine ligation: fast bioconjugation based on inverse-electron-demand Diels-Alder reactivity. J. Am. Chem. Soc. 130, 13518–13519 (2008).An early example of inverse electron demand Diels-Alder reactions developed for the modification of proteins; a reaction that appears to be one of the most rapid in protein modification contexts.
Devaraj, N. K., Weissleder, R. & Hilderbrand, S. Tetrazine-based cycloadditions: application to pretargeted live cell imaging. Bioconjug. Chem. 19, 2297–2299 (2008).
Taylor, M. T., Blackman, M. L., Dmitrenko, O. & Fox, J. M. Design and synthesis of highly reactive dienophiles for the for the tetrazine- trans-cyclooctene ligation. J. Am. Chem. Soc. 133, 9646–9649 (2011).
Devaraj, N. K. & Weissleder, R. Biomedical applications of tetrazine cycloadditions. Acc. Chem. Res. 44, 816–827 (2011).
Seitchik, J. L. et al. Genetically encoded tetrazine amino acid directs rapid site-specific in vivo bioorthogonal ligation with trans-cyclooctenes. J. Am. Chem. Soc. 134, 2898–2901 (2012).
Lang, K. et al. Genetically encoded norbornene directs site-specific cellular protein labelling via a rapid bioorthogonal reaction. Nat. Chem. 4, 298–304 (2012).
Plass, T. et al. Amino acids for Diels-Alder reactions in living cells. Angew. Chem. Int. Ed. 51, 4166–4170 (2012).
Kaya, E. et al. A genetically encoded norbornene amino acid for the mild and selective modification of proteins in a copper-free click reaction. Angew. Chem. Int. Ed. 51, 4466–4469 (2012).
Liang, Y., Mackey, J. L., Lopez, S. a., Liu, F. & Houk, K. N. Control and design of mutual orthogonality in bioorthogonal cycloadditions. J. Am. Chem. Soc. 134, 17904–17907 (2012).
Karver, M. R., Weissleder, R. & Hilderbrand, S. A. Bioorthogonal reaction pairs enable simultaneous, selective, multi-target imaging. Angew. Chem. Int. Ed. 51, 920–922 (2012).
Song, W., Wang, Y., Qu, J., Madden, M. M. & Lin, Q. A photoinducible 1,3-dipolar cycloaddition reaction for rapid, selective modification of tetrazole-containing proteins. Angew. Chem. Int. Ed. 47, 2832–2835 (2008).
Song, W., Wang, Y., Qu, J. & Lin, Q. Selective functionalization of a genetically encoded alkene-containing protein via “ photoclick chemistry ” in bacterial cells. J. Am. Chem. Soc. 130, 9654–9655 (2008).
Yu, Z., Pan, Y., Wang, Z., Wang, J. & Lin, Q. Genetically encoded cyclopropene directs rapid, photoclick-chemistry-mediated protein labeling in mammalian cells. Angew. Chem. Int. Ed. 51, 10600–10604 (2012).
Wang, J. et al. A biosynthetic route to photoclick chemistry on proteins. J. Am. Chem. Soc. 132, 14812–14818 (2010).
Antos, J. M. & Francis, M. B. Transition metal catalyzed methods for site-selective protein modification. Curr. Opin. Chem. Biol. 10, 253–262 (2006).
Dibowski, H. & Schmidtchen, F. P. Bioconjugation of peptides by palladium catalyzed C-C cross-coupling in water. Angew. Chem. Int. Ed. 37, 476–478 (1998).
Ojida, A., Tsutsumi, H., Kasagi, N. & Hamachi, I. Suzuki coupling for protein modification. Tetrahedron Lett. 46, 3301–3305 (2005).
Santoro, S. W., Wang, L., Herberich, B., King, D. S. & Schultz, P. G. An efficient system for the evolution of aminoacyl-tRNA synthetase specificity. Nat. Biotechnol. 20, 1044–1048 (2002).
Kodama, K. et al. Regioselective carbon-carbon bond formation in proteins with palladium catalysis; new protein chemistry by organometallic chemistry. Chembiochem 7, 134–139 (2006).
Kodama, K. et al. Site-specific functionalization of proteins by organopalladium reactions. Chembiochem 8, 232–238 (2007).
Brustad, E. et al. A genetically encoded boronate-containing amino acid. Angew. Chem. Int. Ed. 47, 8220–8223 (2008).
Chalker, J. M., Wood, C. S. C. & Davis, B. G. A convenient catalyst for aqueous and protein Suzuki-Miyaura cross-coupling. J. Am. Chem. Soc. 131, 16346–16347 (2009).A highly biocompatible ligand system developed for the efficient modification of proteins by palladium-mediated C–C bond formation, allowing application of one of the most prevalent reactions in Organic Chemistry to Biology.
Spicer, C. D. & Davis, B. G. Palladium-mediated site-selective Suzuki-Miyaura protein modification at genetically encoded aryl halides. Chem. Commun. 47, 1698–1700 (2011).
Wang, Y.-S. et al. The de novo engineering of pyrrolysyl-tRNA synthetase for genetic incorporation of L-phenylalanine and its derivatives. Mol. Biosyst. 7, 714–717 (2011).
Spicer, C. D., Triemer, T. & Davis, B. G. Palladium-mediated cell-surface labeling. J. Am. Chem. Soc. 134, 800–803 (2012).
Spicer, C. D. & Davis, B. G. Rewriting the bacterial glycocalyx via Suzuki-Miyaura cross-coupling. Chem. Commun. 49, 2747–2749 (2013).
Li, N., Lim, R. K. V., Edwardraja, S. & Lin, Q. Copper-free Sonogashira cross-coupling for functionalization of alkyne-encoded proteins in aqueous medium and in bacterial cells. J. Am. Chem. Soc. 133, 15316–15319 (2011).
Dumas, A. et al. Self-liganded Suzuki-Miyaura coupling for site-selective protein PEGylation. Angew. Chem. Int. Ed. 52, 3916–3921 (2013).
Gao, Z., Gouverneur, V. & Davis, B. G. Enhanced aqueous Suzuki–Miyaura coupling allows site-specific polypeptide 18F-labeling. J. Am. Chem. Soc. 135, 13612–13615 (2013).
Li, J. et al. Ligand-free palladium-mediated site-specific protein labeling inside gram-negative bacterial pathogens. J. Am. Chem. Soc. 135, 7330–7338 (2013).
Lin, Y. A., Chalker, J. M., Floyd, N., Bernardes, G. J. L. & Davis, B. G. Allyl sulfides are privileged substrates in aqueous cross-metathesis: application to site-selective protein modification. J. Am. Chem. Soc. 130, 9642–9643 (2008).
Lin, Y. A. & Davis, B. G. The allylic chalcogen effect in olefin metathesis. Beilstein J. Org. Chem. 6, 1219–1228 (2010).
Chalker, J. M., Lin, Y. A., Boutureira, O. & Davis, B. G. Enabling olefin metathesis on proteins: chemical methods for installation of S-allyl cysteine. Chem. Commun. 3714–3716 (2009).
Lin, Y. A., Chalker, J. M. & Davis, B. G. Olefin cross-metathesis on proteins: investigation of allylic chalcogen effects and guiding principles in metathesis partner selection. J. Am. Chem. Soc. 132, 16805–16811 (2010).
Lin, Y. A. et al. Rapid cross metathesis for protein modifications via chemical access to se-allyl selenocysteine in proteins. J. Am. Chem. Soc. 135, 12156–12159 (2013).
Antos, J. M. & Francis, M. B. Selective tryptophan modification with rhodium carbenoids in aqueous solution. J. Am. Chem. Soc. 126, 10256–10257 (2004).
Antos, J. M., McFarland, J. M., Iavarone, A. T. & Francis, M. B. Chemoselective tryptophan labeling with rhodium carbenoids at mild pH. J. Am. Chem. Soc. 131, 6301–6308 (2009).
Popp, B. V. & Ball, Z. T. Structure-selective modification of aromatic side chains with dirhodium metallopeptide catalysts. J. Am. Chem. Soc. 132, 6660–6662 (2010).
Chen, Z. et al. Catalytic protein modification with dirhodium metallopeptides: specificity in designed and natural systems. J. Am. Chem. Soc. 134, 10138–10145 (2012).
Alam, J., Keller, T. H. & Loh, T.-P. Functionalization of peptides and proteins by Mukaiyama aldol reaction. J. Am. Chem. Soc. 132, 9546–9548 (2010).
Han, M.-J., Xiong, D.-C. & Ye, X.-S. Enabling Wittig reaction on site-specific protein modification. Chem. Commun. 48, 11079–11081 (2012).
Wang, L., Zhang, Z., Brock, A. & Schultz, P. G. Addition of the keto functional group to the genetic code of Escherichia coli. Proc. Natl Acad. Sci. USA 100, 56–61 (2003).
Dirksen, A. & Dawson, P. E. Rapid oxime and hydrazone ligations with aromatic aldehydes for biomolecular labeling. Bioconjug. Chem. 19, 2543–2548 (2008).
Geoghegan, K. F. & Stroh, J. G. Site-directed conjugation of nonpeptide groups to peptides and proteins via periodate oxidation of a 2-amino alcohol. Application to modification at N-terminal serine. Bioconjug. Chem. 3, 138–146 (1992).
Gilmore, J. M., Scheck, R. A., Esser-Kahn, A. P., Joshi, N. S. & Francis, M. B. N-terminal protein modification through a biomimetic transamination reaction. Angew. Chem. Int. Ed. 45, 5307–5311 (2006).
Scheck, R. A., Dedeo, M. T., Iavarone, A. T. & Francis, M. B. Optimization of a biomimetic transamination reaction. J. Am. Chem. Soc. 130, 11762–11770 (2008).
Scheck, R. A. & Francis, M. B. Regioselective labeling of antibodies through N-terminal transamination. ACS Chem. Biol. 2, 247–251 (2007).
Carrico, Z. M. et al. Terminal labeling of filamentous phage to create cancer marker imaging agents. ACS Nano 6, 6675–6680 (2012).
Cornish, V. W., Hahn, K. M. & Schultz, P. G. Site-specific protein modification using a ketone handle. J. Am. Chem. Soc. 118, 8150–8151 (1996).
Zhang, Z. et al. A new strategy for the site-specific modification of proteins in vivo. Biochemistry 42, 6735–6746 (2003).
Chin, J. W. et al. An expanded eukaryotic genetic code. Science 301, 964–967 (2003).
Huang, Y. et al. Genetic incorporation of an aliphatic keto-containing amino acid into proteins for their site-specific modifications. Bioorg. Med. Chem. Lett. 20, 878–880 (2010).
Zeng, H., Xie, J. & Schultz, P. G. Genetic introduction of a diketone-containing amino acid into proteins. Bioorg. Med. Chem. Lett. 16, 5356–5359 (2006).
Carrico, I. S., Carlson, B. L. & Bertozzi, C. R. Introducing genetically encoded aldehydes into proteins. Nat. Chem. Biol. 3, 321–322 (2007).
Liu, H., Wang, L., Brock, A., Wong, C.-H. & Schultz, P. G. A method for the generation of glycoprotein mimetics. J. Am. Chem. Soc. 125, 1702–1703 (2003).
Ye, S. et al. Site-specific incorporation of keto amino acids into functional G protein-coupled receptors using unnatural amino acid mutagenesis. J. Biol. Chem. 283, 1525–1533 (2008).
Hutchins, B. M. et al. Site-specific coupling and sterically controlled formation of multimeric antibody fab fragments with unnatural amino acids. J. Mol. Biol. 406, 595–603 (2011).
Hudak, J. E. et al. Synthesis of heterobifunctional protein fusions using copper-free click chemistry and the aldehyde tag. Angew. Chem. Int. Ed. 51, 4161–4165 (2012).
Kim, C. H. et al. Synthesis of bispecific antibodies using genetically encoded unnatural amino acids. J. Am. Chem. Soc. 134, 9918–9921 (2012).
Brustad, E. M., Lemke, E., Schultz, P. G. & Deniz, A. A general and efficient method for the site-specific dual-labeling of proteins for single molecule fluorescence resonance energy transfer. J. Am. Chem. Soc. 130, 17664–17665 (2008).
Sasaki, T., Kodama, K., Suzuki, H., Fukuzawa, S. & Tachibana, K. N-terminal labeling of proteins by the Pictet-Spengler reaction. Bioorg. Med. Chem. Lett. 18, 4550–4553 (2008).
Agarwal, P., Weijden, J. V. D., Sletten, E. M., Rabuka, D. & Bertozzi, C. R. A Pictet-Spengler ligation for protein chemical modification. Proc. Natl Acad. Sci. USA 110, 46–51 (2012).
Wang, J., Schiller, S. M. & Schultz, P. G. A biosynthetic route to dehydroalanine-containing proteins. Angew. Chem. Int. Ed. 119, 6973–6975 (2007).
Guo, J., Wang, J., Lee, J. S. & Schultz, P. G. Site-specific incorporation of methyl- and acetyl-lysine analogues into recombinant proteins. Angew. Chem. Int. Ed. 120, 6499–6501 (2008).
Bernardes, G. J. L., Chalker, J. M., Errey, J. C. & Davis, B. G. Facile conversion of cysteine and alkyl cysteines to dehydroalanine on protein surfaces: versatile and switchable access to functionalized proteins. J. Am. Chem. Soc. 130, 5052–5053 (2008).
Chalker, J. M. et al. Methods for converting cysteine to dehydroalanine on peptides and proteins. Chem. Sci. 2, 1666–1676 (2011).
Chalker, J. M., Lercher, L., Rose, N. R., Schofield, C. J. & Davis, B. G. Conversion of cysteine into dehydroalanine enables access to synthetic histones bearing diverse post-translational modifications. Angew. Chem. Int. Ed. 51, 1835–1839 (2012).
Wang, Z. U. et al. A facile method to synthesize histones with posttranslational modification mimics. Biochemistry 51, 5232–5234 (2012).
Floyd, N., Vijayakrishnan, B., Koeppe, J. R. & Davis, B. G. Thiyl glycosylation of olefinic proteins: S-linked glycoconjugate synthesis. Angew. Chem. Int. Ed. 48, 7798–7802 (2009).
Sletten, E. M. & Bertozzi, C. R. From mechanism to mouse: a tale of two bioorthgonal reactions. Acc. Chem. Res. 44, 666–676 (2011).
Chalker, J. M., Bernardes, J. L., Davis, B. G. & Bernardes, G. J. L. A "tag-and-modify" approach to site-selective protein modification. Acc. Chem. Res. 44, 730–741 (2011).
Chen, Y.-X., Triola, G. & Waldmann, H. Bioorthognal chemistry for site-specific labeling and surface immobilization of proteins. Acc. Chem. Res. 44, 762–773 (2011).
Moore, J. E. & Ward, W. H. Cross-linking of bovine plasma albumin and wool keratin. J. Am. Chem. Soc. 78, 2414–2418 (1948).
Seim, K. L., Obermeyer, A. C. & Francis, M. B. Oxidative modification of native protein residues using cerium(IV) ammonium nitrate. J. Am. Chem. Soc. 133, 16970–16976 (2011).
Schlick, T. L., Ding, Z., Kovacs, E. W. & Francis, M. B. Dual-surface modification of the tobacco mosaic virus. J. Am. Chem. Soc. 127, 3718–3723 (2005).
McFarland, J. M., Joshi, N. S. & Francis, M. B. Characterization of a three-component coupling reaction on proteins by isotopic labeling and nuclear magnetic resonance spectroscopy. J. Am. Chem. Soc. 130, 7639–7644 (2008).
Tilley, S. D. & Francis, M. B. Tyrosine-selective protein alkylation using pi-allylpalladium complexes. J. Am. Chem. Soc. 128, 1080–1081 (2006).
Espuña, G. et al. Iodination of proteins by IPy2BF4, a new tool in protein chemistry. Biochemistry 4, 5957–5963 (2006).
Hooker, J. M., Esser-Kahn, A. P. & Francis, M. B. Modification of aniline containing proteins using an oxidative coupling strategy. J. Am. Chem. Soc. 128, 15558–15559 (2006).
Carrico, Z. M., Romanini, D. W., Mehl, R. & Francis, M. B. Oxidative coupling of peptides to a virus capsid containing unnatural amino acids. Chem. Commun. 1205–1207 (2008).
Tong, G. J., Hsiao, S. C., Carrico, Z. M. & Francis, M. B. Viral capsid DNA aptamer conjugates as multivalent cell-targeting vehicles. J. Am. Chem. Soc. 131, 11174–11178 (2009).
Wittrock, S., Becker, T. & Kunz, H. Synthetic vaccines of tumor-associated glycopeptide antigens by immune-compatible thioether linkage to bovine serum albumin. Angew. Chem. Int. Ed. 46, 5226–5230 (2007).
Li, Y. et al. Genetically encoded alkenyl–pyrrolysine analogues for thiol–ene reaction mediated site-specific protein labeling. Chem. Sci. 3, 2766–2766 (2012).
Dondoni, A., Massi, A., Nanni, P. & Roda, A. A new ligation strategy for peptide and protein glycosylation: photoinduced thiol-ene coupling. Chem. Eur. J. 15, 11444–11449 (2009).
Li, Y., Pan, M., Li, Y., Huang, Y. & Guo, Q. Thiol-yne radical reaction mediated site-specific protein labeling via genetic incorporation of an alkynyl-L-lysine analogue. Org. Biomol. Chem. 11, 2624–2629 (2013).
Fleet, G. W. J. & Porter, R. R. Affinity labelling of antibodies with the aryl nitrene as reactive group. Nature 224, 511–512 (1969).
Chin, J. W., Martin, A. B., King, D. S., Wang, L. & Schultz, P. G. Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli. Proc. Natl Acad. Sci. USA 99, 11020–11024 (2002).
Chin, J. W. et al. Addition of p-azido-L-phenylalanine to the genetic code of Escherichia coli. J. Am. Chem. Soc. 124, 9026–9027 (2002).
Tippmann, E. M., Liu, W., Summerer, D., Mack, A. V. & Schultz, P. G. A genetically encoded diazirine photocrosslinker in Escherichia coli. Chembiochem 8, 2210–2214 (2007).
Chou, C., Uprety, R., Davis, L., Chin, J. W. & Deiters, A. Genetically encoding an aliphatic diazirine for protein photocrosslinking. Chem. Sci. 2, 480–480 (2011).
Griffin, B. A., Adams, S. R. & Tsien, R. Y. Specific covalent labeling of recombinant protein molecules inside live cells. Science 281, 269–272 (1998).
Adams, S. R. et al. New biarsenical ligands and tetracysteine motifs for protein labeling in vitro and in vivo: synthesis and biological applications. J. Am. Chem. Soc. 124, 6063–6076 (2002).
Tsukiji, S., Miyagawa, M., Takaoka, Y., Tamura, T. & Hamachi, I. Ligand-directed tosyl chemistry for protein labeling in vivo. Nat. Chem. Biol. 5, 341–343 (2009).A thought-provoking illustration that enhanced inherent or situation-dependent selectivity can be achieved in convergent protein alteration through a range of mechanisms and using even simple reactions.
Tamura, T., Tsukiji, S. & Hamachi, I. Native FKBP12 engineering by ligand-directed tosyl chemistry: labeling properties and application to photo-cross-linking of protein complexes in vitro and in living cells. J. Am. Chem. Soc. 134, 2216–2226 (2012).
Wang, L., Brock, A., Herberich, B. & Schultz, P. G. Expanding the genetic code of Escherichia coli. Science 292, 498–500 (2001).Unnatural amino acids incorporated site-selectively into proteins by amber codon reassignment and suppression has proven to be a lynchpin technique for enabling the creation of substrates for site-selective protein chemistry.
Liu, C. C. & Schultz, P. G. Adding new chemistries to the genetic code. Annu. Rev. Biochem. 79, 413–444 (2010).
Greiss, S. & Chin, J. W. Expanding the genetic code of an animal. J. Am. Chem. Soc. 133, 14196–14199 (2011).
Parrish, A. R. et al. Expanding the genetic code of Caenorhabditis elegans using bacterial aminoacyl-tRNA synthetase/tRNA pairs. ACS Chem. Biol. 7, 1292–1302 (2012).
Johnson, D. B. F. et al. Release factor one is nonessential in Escherichia coli. ACS Chem. Biol. 7, 1337–1344 (2012).
Isaacs, F. J. et al. Precise manipulation of chromosomes in vivo enables genome-wide codon replacement. Science 333, 348–353 (2011).
Acknowledgements
We would like to thank UCB and the BBSRC for funding (CDS studentship). B.G.D. is a Royal Society Wolfson Research Merit Award Recipient.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article

Cite this article
Spicer, C., Davis, B. Selective chemical protein modification. Nat Commun 5, 4740 (2014). https://doi.org/10.1038/ncomms5740
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms5740
This article is cited by
-
Selenium chemistry for spatio-selective peptide and protein functionalization
Nature Reviews Chemistry (2024)
-
Location-agnostic site-specific protein bioconjugation via Baylis Hillman adducts
Nature Communications (2024)
-
Electrochemical labelling of hydroxyindoles with chemoselectivity for site-specific protein bioconjugation
Nature Chemistry (2024)
-
Oxidative cyclization reagents reveal tryptophan cation–π interactions
Nature (2024)
-
Membrane linked RNA glycosylation as new trend to envision epi-transcriptome epoch
Cancer Gene Therapy (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
