
Featured
-
-
Article
| Open Access Robust single-cell DNA methylome profiling with snmC-seq2
Robust single-cell DNA methylome profiling with snmC-seq2
Single-cell DNA methylome profiling allows the study of epigenomic heterogeneity in tissues but has been impeded by library quality. Here the authors demonstrate snmC-seq2 which improves mapping, throughput and library complexity.
- Chongyuan Luo
- , Angeline Rivkin
- & Joseph R. Ecker
-
Article
| Open Access Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer
Differentiation-state plasticity is a targetable resistance mechanism in basal-like breast cancer
Resistance to therapy can be driven by intratumoral heterogeneity. Here, the authors show that drug tolerant persistent cell populations emerge during treatment, and these emergent populations arise through epigenetically mediated cell state transitions rather than sub population selection.
- Tyler Risom
- , Ellen M. Langer
- & Rosalie C. Sears
-
Article
| Open Access Autosomal genetic variation is associated with DNA methylation in regions variably escaping X-chromosome inactivation
Autosomal genetic variation is associated with DNA methylation in regions variably escaping X-chromosome inactivation
DNA methylation is critically involved in X chromosome inactivation (XCI) and dosage compensation, yet some X-chromosomal genes escape XCI. Here, Lujik et al. identify three autosomal genetic loci that associate with differential DNA methylation near genes that variably escape XCI in females.
- René Luijk
- , Haoyu Wu
- & Bastiaan T. Heijmans
-
Article
| Open Access High-throughput chromatin accessibility profiling at single-cell resolution
High-throughput chromatin accessibility profiling at single-cell resolution
Single-cell chromatin accessibility is a promising means to identify regulatory programs in mixtures of cells. Here the authors describe µ-ATAC-seq, a low-cost method that can generate thousands of accessibility profiles per day.
- Anja Mezger
- , Sandy Klemm
- & William Greenleaf
-
Article
| Open Access Epigenetic dysregulation of naive CD4+ T-cell activation genes in childhood food allergy
Epigenetic dysregulation of naive CD4+ T-cell activation genes in childhood food allergy
Immunoglobulin E (IgE)-mediated food allergy is a major issue that affects 2–10% of infants. Here the authors study the epigenetic regulation of the naive CD4+ T cell activation response among children with IgE-mediated food allergy finding epigenetic dysregulation in the early stages of signal transduction through the T cell receptor complex.
- David Martino
- , Melanie Neeland
- & Richard Saffery
-
Article
| Open Access Pan-cancer deconvolution of tumour composition using DNA methylation
Pan-cancer deconvolution of tumour composition using DNA methylation
Determining the extent of immune cell infiltration into solid tumours is essential for adequate therapeutic response. Here the authors develop a DNA methylation-based approach for tumour cell fraction deconvolution and analyse tumour composition and genomics across a wide spectrum of solid cancers.
- Ankur Chakravarthy
- , Andrew Furness
- & Tim R. Fenton
-
Article
| Open Access Epigenome-wide DNA methylation profiling in Progressive Supranuclear Palsy reveals major changes at DLX1
Epigenome-wide DNA methylation profiling in Progressive Supranuclear Palsy reveals major changes at DLX1
Progressive supranuclear palsy (PSP) is a neurodegenerative disease characterized by aggregation of Tau, encoded by MAPT. Here, the authors perform an EWAS for PSP in prefrontal lobe tissue and find hypermethylation of DLX1 and its antisense transcript DLX1AS to associate with MAPT expression.
- Axel Weber
- , Sigrid C. Schwarz
- & Ulrich Müller
-
Article
| Open Access Enhancer histone-QTLs are enriched on autoimmune risk haplotypes and influence gene expression within chromatin networks
Enhancer histone-QTLs are enriched on autoimmune risk haplotypes and influence gene expression within chromatin networks
Disease risk variants can exert their influence on phenotypes by altering epigenome function. Here, Pelikan et al. show that variants inducing allelic imbalance in histone marks in lymphoblastoid cell lines from lupus patients are enriched in autoimmune disease haplotypes and influence gene expression.
- Richard C. Pelikan
- , Jennifer A. Kelly
- & Patrick M. Gaffney
-
Article
| Open Access DNA methylation as a mediator of HLA-DRB1*15:01 and a protective variant in multiple sclerosis
DNA methylation as a mediator of HLA-DRB1*15:01 and a protective variant in multiple sclerosis
The human leukocyte antigen (HLA) haplotype DRB1*15:01 is the major risk factor for multiple sclerosis (MS). Here the authors find that DNA methylation at HLA-DRB1 gene mediates the effect of DRB1*15:01 and of a protective HLA variant on HLA-DRB1 expression and the risk of MS.
- Lara Kular
- , Yun Liu
- & Maja Jagodic
-
Article
| Open Access Identification of rare de novo epigenetic variations in congenital disorders
Identification of rare de novo epigenetic variations in congenital disorders
A proportion of neurodevelopmental disorder and congenital anomaly cases remain without a genetic diagnosis. Here, the authors study aberrations of DNA methylation in such cases and find that epivariations might provide an explanation for some of these undiagnosed patients.
- Mafalda Barbosa
- , Ricky S. Joshi
- & Andrew J. Sharp
-
Article
| Open Access Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes
Distinct epigenetic landscapes underlie the pathobiology of pancreatic cancer subtypes
Pancreatic ductal adenocarcinoma (PDAC) is a complex disease and its underlying epigenomic heterogeneity is not fully understood. Here, the authors utilize patient-derived PDAC xenografts to define the epigenomic landscape of PDAC, highlighting chromatin states linked to differing disease aggressiveness and survival.
- Gwen Lomberk
- , Yuna Blum
- & Raul Urrutia
-
Article
| Open Access Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width
Methionine metabolism influences genomic architecture and gene expression through H3K4me3 peak width
Methionine availability is known to affect the global levels of histone methylation. Here the authors investigate the metabolically driven dynamics of H3K4me3 and find that methionine availability influences peak width, which is linked to changes in expression of genes associated with cell fate and cancer.
- Ziwei Dai
- , Samantha J. Mentch
- & Jason W. Locasale
-
Article
| Open Access Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes
Comprehensive epigenetic landscape of rheumatoid arthritis fibroblast-like synoviocytes
Fibroblast-like synoviocytes (FLS) in the intimal layer of the synovium can become invasive and destroy cartilage in patients with rheumatoid arthritis (RA). Here the authors integrate a variety of epigenomic data to map the epigenome of FLS in RA and identify potential therapeutic targets.
- Rizi Ai
- , Teresina Laragione
- & Gary S. Firestein
-
Article
| Open Access Changes in genome organization of parasite-specific gene families during the Plasmodium transmission stages
Changes in genome organization of parasite-specific gene families during the Plasmodium transmission stages
The development of malaria parasites is controlled by coordinated changes in gene expression. Here, the authors show that the three-dimensional genome structure of human malaria parasites is strongly connected with transcriptional activity of specific gene families throughout the life cycles of Plasmodium falciparum and Plasmodium vivax parasites.
- Evelien M. Bunnik
- , Kate B. Cook
- & Karine G. Le Roch
-
Article
| Open Access B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation
B cell activation and plasma cell differentiation are inhibited by de novo DNA methylation
DNA methylation is known to contribute to B cell differentiation, but de novo methylation has not been studied in this context. Here the authors use a conditional Dnmt3a/b knockout mouse to map the function of de novo DNA methylation in B cell differentiation and the development of humoral immunity.
- Benjamin G. Barwick
- , Christopher D. Scharer
- & Jeremy M. Boss
-
Article
| Open Access Snord116-dependent diurnal rhythm of DNA methylation in mouse cortex
Snord116-dependent diurnal rhythm of DNA methylation in mouse cortex
Many genes have oscillating gene expression pattern in circadian centers of the brain. This study shows cortical diurnal DNA methylation oscillation in a mouse model of Prader-Willi syndrome, and describes corresponding changes in gene expression and chromatin compaction.
- Rochelle L. Coulson
- , Dag H. Yasui
- & Janine M. LaSalle
-
Article
| Open Access PREDICTD PaRallel Epigenomics Data Imputation with Cloud-based Tensor Decomposition
PREDICTD PaRallel Epigenomics Data Imputation with Cloud-based Tensor Decomposition
Assays to characterize the epigenome and interrogate chromatin state genome wide have so far been performed in a selected set of conditions. Here, Durham et al. develop a computational method based on tensor decomposition to impute missing experiments in collections of epigenomics experiments.
- Timothy J. Durham
- , Maxwell W. Libbrecht
- & William Stafford Noble
-
Article
| Open Access ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration
ATAC-Seq analysis reveals a widespread decrease of chromatin accessibility in age-related macular degeneration
Age-related macular degeneration (AMD) leads to dysfunctional retinal pigment epithelium (RPE) and vision loss. Here, the authors perform ATAC-seq to study chromatin accessibility and find that differentially accessible regions are enriched for photoreceptor and RPE-specific transcription factors in AMD
- Jie Wang
- , Cristina Zibetti
- & Jiang Qian
-
Article
| Open Access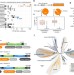 Recurrent acquisition of cytosine methyltransferases into eukaryotic retrotransposons
Recurrent acquisition of cytosine methyltransferases into eukaryotic retrotransposons
Cytosine methyltransferases (DNMTs) often silence transposons in eukaryotic genomes. Here the authors describe the recurrent acquisition of DNMTs by transposons from two distantly-related eukaryotes and suggest that methylation of CG dinucleotides by transposon DNMTs could modify the host epigenome in dinoflagellates.
- Alex de Mendoza
- , Amandine Bonnet
- & Ryan Lister
-
Article
| Open Access Atrx inactivation drives disease-defining phenotypes in glioma cells of origin through global epigenomic remodeling
Atrx inactivation drives disease-defining phenotypes in glioma cells of origin through global epigenomic remodeling
ATRX inactivation frequently occurs in glioma. Here, the authors explore the role of ATRX inactivation in oncogenesis, highlighting ATRX deficiency driven epigenomic changes that influence the expression of genes crucial to the oncogenic phenotype.
- Carla Danussi
- , Promita Bose
- & Jason T. Huse
-
Article
| Open Access Dissecting super-enhancer hierarchy based on chromatin interactions
Dissecting super-enhancer hierarchy based on chromatin interactions
Super-enhancers (SEs) are important regulatory elements for gene expression, but their intrinsic properties remain poorly understood. Here the authors analyse Hi-C and ChIP-seq data and find that a significant fraction of SEs are hierarchically organized, containing both hub and non-hub enhancers.
- Jialiang Huang
- , Kailong Li
- & Guo-Cheng Yuan
-
Article
| Open Access Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits
Integrative analysis of omics summary data reveals putative mechanisms underlying complex traits
The identification of the causal gene at a GWAS locus remains to be a challenging task. Here, using the SMR & HEIDI method to integrate GWAS, eQTL and mQTL data, Wu et al. map DNA methylation sites to the transcriptome and thereby prioritize functionally relevant genes for 12 human complex traits.
- Yang Wu
- , Jian Zeng
- & Jian Yang
-
Article
| Open Access TET-mediated epimutagenesis of the Arabidopsis thaliana methylome
TET-mediated epimutagenesis of the Arabidopsis thaliana methylome
In plants, variation in DNA methylation can influence agronomically important traits. Here the authors show that expression of a human Ten-eleven translocation enzyme can generate heritable hypomethylation alleles in Arabidopsis, providing a novel method to manipulate DNA methylation for breeding and research purposes.
- Lexiang Ji
- , William T. Jordan
- & Robert J. Schmitz
-
Article
| Open Access Sheep genome functional annotation reveals proximal regulatory elements contributed to the evolution of modern breeds
Sheep genome functional annotation reveals proximal regulatory elements contributed to the evolution of modern breeds
The domestication of plants and animals causes genomic changes underlying various morphologic, physiologic and behavioral changes. Here, Naval-Sanchez et al. provide a ChIP-Seq validated comparative functional annotation of the sheep genome, and show widespread evolution of proximal regulatory elements.
- Marina Naval-Sanchez
- , Quan Nguyen
- & James Kijas
-
Article
| Open Access Co-occurring expression and methylation QTLs allow detection of common causal variants and shared biological mechanisms
Co-occurring expression and methylation QTLs allow detection of common causal variants and shared biological mechanisms
Most expression QTLs (eQTLs) co-occur with a DNA methylation QTL (meQTL), suggesting a common causal variant. Here the authors analyse DNA and RNA from blood and identify eQTL-meQTL pairs likely to share a causal variant, finding that expression and methylation are often genetically co-regulated.
- Brandon L. Pierce
- , Lin Tong
- & Habibul Ahsan
-
Article
| Open Access Exploiting genetic variation to uncover rules of transcription factor binding and chromatin accessibility
Exploiting genetic variation to uncover rules of transcription factor binding and chromatin accessibility
Single nucleotide variants (SNVs) can affect chromatin occupancy by transcription factors (TF). Here the authors mine naturally occurring SNVs to probe cis elements and define contextual sequences that govern in vivo transcription factor chromatin occupancy and chromatin accessibility.
- Vivek Behera
- , Perry Evans
- & Gerd A. Blobel
-
Article
| Open Access Genome-wide tracking of dCas9-methyltransferase footprints
Genome-wide tracking of dCas9-methyltransferase footprints
Catalytically inactive Cas9 fused to a methyltransferase has emerged as a promising epigenome modifying tool. Here the authors generate a methylation depleted but maintenance competent mouse ES cell line and find ubiquitous off-target activity.
- Christina Galonska
- , Jocelyn Charlton
- & Alexander Meissner
-
Article
| Open Access Stratification of TAD boundaries reveals preferential insulation of super-enhancers by strong boundaries
Stratification of TAD boundaries reveals preferential insulation of super-enhancers by strong boundaries
Topologically associating domains (TADs) detected by Hi-C technologies are megabase-scale areas of highly interacting chromatin. Here Gong, Lazaris et al. develop a computational approach to improve the reproducibility of Hi-C contact matrices and stratify TAD boundaries based on their insulating strength.
- Yixiao Gong
- , Charalampos Lazaris
- & Aristotelis Tsirigos
-
Article
| Open Access Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo
Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo
How the cardiac myocyte epigenome is rearranged during development, postnatal maturation and disease is not well understood. Here, the authors investigate the human cardiac myocyte epigenome during development and chronic heart failure and identify distinct epigenetic programs regulating these processes.
- Ralf Gilsbach
- , Martin Schwaderer
- & Lutz Hein
-
Article
| Open Access Environmental enrichment increases transcriptional and epigenetic differentiation between mouse dorsal and ventral dentate gyrus
Environmental enrichment increases transcriptional and epigenetic differentiation between mouse dorsal and ventral dentate gyrus
Environmental enrichment has functional and molecular effects on mammalian hippocampus. Here, Zhang and colleagues show that environmental enrichment of mice is correlated with dorsal-ventral asymmetry in transcription and DNA methylation of the dentate gyrus.
- Tie-Yuan Zhang
- , Christopher L. Keown
- & Michael J. Meaney
-
Article
| Open Access AICDA drives epigenetic heterogeneity and accelerates germinal center-derived lymphomagenesis
AICDA drives epigenetic heterogeneity and accelerates germinal center-derived lymphomagenesis
In diffuse large B-cell lymphoma (DLBCL) increased epigenetic heterogeneity in the form of cytosine methylation is known to link to a poor clinical outcome. Here, the authors show that AICDA, an enzyme required for DLBCL pathogenesis, increases cytosine methylation heterogeneity.
- Matt Teater
- , Pilar M. Dominguez
- & Rita Shaknovich
-
Article
| Open Access Obligatory and facilitative allelic variation in the DNA methylome within common disease-associated loci
Obligatory and facilitative allelic variation in the DNA methylome within common disease-associated loci
Genomic polymorphisms affect the epigenome, which in turn influences how epigenome- and genome-wide analysis are interpreted. Here, the authors characterise allelic differences in DNA methylation driven by obligatory or facilitative genetic effects, which may affect disease-related loci.
- Christopher G. Bell
- , Fei Gao
- & Timothy D. Spector
-
Article
| Open Access DNA methylation signatures of illicit drug injection and hepatitis C are associated with HIV frailty
DNA methylation signatures of illicit drug injection and hepatitis C are associated with HIV frailty
Intravenous illicit drug use (IDU) and hepatitis C infection (HCV) often occur among HIV-infected individuals. Here, the authors report an epigenome-wide association analysis of IDU and HCV in HIV-infected individuals, finding that their associated methylation signatures inform HIV frailty.
- Xinyu Zhang
- , Ying Hu
- & Ke Xu
-
Article
| Open Access RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia
RAS-pathway mutation patterns define epigenetic subclasses in juvenile myelomonocytic leukemia
Juvenile myelomonocytic leukemia (JMML) is an aggressive disease with limited options for treatment. Here, the authors analyse the DNA methylome and mutational profile of JMML to define three subgroups with unique molecular and clinical characteristics.
- Daniel B. Lipka
- , Tania Witte
- & Christoph Plass
-
Article
| Open Access Evolution of sequence-specific anti-silencing systems in Arabidopsis
Evolution of sequence-specific anti-silencing systems in Arabidopsis
Eukaryotes often silence transposable elements (TEs) via DNA methylation. Here, the authors show that evolution of VANC, an Arabidopsis anti-silencing factor, and its target motifs allows sequence-specific demethylation, suggesting a way TEs can proliferate while minimizing damage to the host genome.
- Aoi Hosaka
- , Raku Saito
- & Tetsuji Kakutani
-
Article
| Open Access Single-cell absolute contact probability detection reveals chromosomes are organized by multiple low-frequency yet specific interactions
Single-cell absolute contact probability detection reveals chromosomes are organized by multiple low-frequency yet specific interactions
Eukaryotic genomes are partitioned into self-interacting modules or topologically associated domains (TADs) that exist at the kilo-megabase scale. Here Cattoni et al. combine super-resolution microscopy with DNA-labeling methods to quantify absolute frequencies of interactions within TADs.
- Diego I. Cattoni
- , Andrés M. Cardozo Gizzi
- & Marcelo Nollmann
-
Article
| Open Access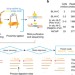 BL-Hi-C is an efficient and sensitive approach for capturing structural and regulatory chromatin interactions
BL-Hi-C is an efficient and sensitive approach for capturing structural and regulatory chromatin interactions
Chromatin interactions and genome architecture are key regulators of gene expression. Here the authors present Bridge-Linker-Hi-C to map active chromatin loops and enhancer-promoter interactions.
- Zhengyu Liang
- , Guipeng Li
- & Yang Chen
-
Article
| Open Access Structural and spatial chromatin features at developmental gene loci in human pluripotent stem cells
Structural and spatial chromatin features at developmental gene loci in human pluripotent stem cells
Higher-order chromatin organization regulates the expression of transcriptional programs that control cell function. Here, the authors show that chromatin interaction profiles and nuclear positions at developmental gene loci differ between human somatic and pluripotent stem cells.
- Hiroki Ikeda
- , Masamitsu Sone
- & Takuya Yamamoto
-
Article
| Open Access Genome-wide mapping of genetic determinants influencing DNA methylation and gene expression in human hippocampus
Genome-wide mapping of genetic determinants influencing DNA methylation and gene expression in human hippocampus
Most SNPs are located in non-coding genomic regions and their function remains elusive. Here, the authors perform a genome-wide scan of expression and DNA methylation quantitative trait loci in human hippocampal tissue to provide a resource for the functional interpretation of SNPs in brain disorders.
- Herbert Schulz
- , Ann-Kathrin Ruppert
- & Sven Cichon
-
Article
| Open Access Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells
Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells
The repertoire of tissue-specific distal regulators of gene transcription enhancers defines homeostasis or disease. Here, the authors reveal the enhancer and super-enhancer signature of vascular smooth muscle cells under normal and angiotensin II stimuli, providing new insight into the transcriptional regulation of vascular pathologies.
- Sadhan Das
- , Parijat Senapati
- & Rama Natarajan
-
Article
| Open Access Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer
Acetylated histone variant H2A.Z is involved in the activation of neo-enhancers in prostate cancer
Acetylation of the histone variant H2A.Z at gene promoters is associated with oncogene activation; however, it is unclear if such modification has a role in regulating the function of enhancers. Here the authors show that acetylated H2A.Z is redistributed at cancer neo-enhancers and regulates the activity of specific enhancers of cancer-related genes.
- Fátima Valdés-Mora
- , Cathryn M. Gould
- & Susan J. Clark
-
Article
| Open Access Genome-wide genetic and epigenetic analyses of pancreatic acinar cell carcinomas reveal aberrations in genome stability
Genome-wide genetic and epigenetic analyses of pancreatic acinar cell carcinomas reveal aberrations in genome stability
Pancreatic acinar cell carcinoma (ACC) is an aggressive exocrine tumor with largely unknown biology. Here, the authors perform genome- and epigenome-wide analyses from normal and ACC pancreatic tissue that identify aberrations in genome stability and cell cycle control.
- Cornelia Jäkel
- , Frank Bergmann
- & Peter Schmezer
-
Article
| Open Access Cross-tissue integration of genetic and epigenetic data offers insight into autism spectrum disorder
Cross-tissue integration of genetic and epigenetic data offers insight into autism spectrum disorder
“There have been a number of recent epigenetic studies on autism spectrum disorder. Here, the authors integrate genetic and epigenetic data from cord and peripheral blood and also from brain tissues to show the potential of blood-based epigenetic data to provide insights into psychiatric disorders.”
- Shan V. Andrews
- , Shannon E. Ellis
- & M. Daniele Fallin
-
Article
| Open Access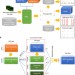 Genome-wide prediction of DNase I hypersensitivity using gene expression
Genome-wide prediction of DNase I hypersensitivity using gene expression
A map of the activities of all genomic regulatory elements across cell types and conditions would be a tremendous resource. The computational method introduced here predicts genome-wide accessible sites from gene expression data and allows the authors to build a database of regulatory element activities using publicly available transcriptome data.
- Weiqiang Zhou
- , Ben Sherwood
- & Hongkai Ji
-
Article
| Open Access Identifying topologically associating domains and subdomains by Gaussian Mixture model And Proportion test
Identifying topologically associating domains and subdomains by Gaussian Mixture model And Proportion test
Spatial organization of the genome plays a crucial role in regulating gene expression. Here the authors introduce GMAP, the Gaussian Mixture model And Proportion test, to identify topologically associating domains and subdomains in Hi-C data.
- Wenbao Yu
- , Bing He
- & Kai Tan
-
Article
| Open Access Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression
Histone variant H2A.J accumulates in senescent cells and promotes inflammatory gene expression
Senescence of mammalian cells is characterized by proliferative arrest and expression of an inflammatory phenotype. Here the authors show the H2A variant H2A.J, found only in mammals, accumulates following persistent DNA damage or natural aging.
- Kévin Contrepois
- , Clément Coudereau
- & Carl Mann
-
Article
| Open Access Multi-scale chromatin state annotation using a hierarchical hidden Markov model
Multi-scale chromatin state annotation using a hierarchical hidden Markov model
The impact of chromatin structure on gene expression makes it integral to our understanding of developmental and disease processes. Here, the authors introduce a hierarchical hidden Markov model to systematically annotate chromatin states at multiple length scales, and demonstrate its utility for the elucidation of the role of chromatin structure in gene expression.
- Eugenio Marco
- , Wouter Meuleman
- & Guo-Cheng Yuan
-
Article
| Open Access Diurnal and seasonal molecular rhythms in human neocortex and their relation to Alzheimer’s disease
Diurnal and seasonal molecular rhythms in human neocortex and their relation to Alzheimer’s disease
Diurnal and seasonal rhythms modulate brain function, but we do not know the genomic basis for these rhythms. Here, Limet al. show diurnal and seasonal rhythms of gene expression in the human brain, their relationship to histone acetylation and DNA methylation, and their disruption in Alzheimer’s disease.
- Andrew S. P. Lim
- , Hans-Ulrich Klein
- & Philip L. De Jager
-
Article
| Open Access DNA methylation signatures in peripheral blood strongly predict all-cause mortality
DNA methylation signatures in peripheral blood strongly predict all-cause mortality
DNA methylation is modulated by environmental factors and has a role in many complex diseases. Here, the authors find that methylation at specific DNA sites is associated with all-cause mortality, and a methylation-based risk score may be informative for risk assessment and stratification.
- Yan Zhang
- , Rory Wilson
- & Hermann Brenner
 Epigenomic map of human liver reveals principles of zonated morphogenic and metabolic control
Epigenomic map of human liver reveals principles of zonated morphogenic and metabolic control