
Article
|
Open Access
Featured
-
-
Article
| Open Access Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives
Inhibition mechanism of SARS-CoV-2 main protease by ebselen and its derivatives
Ebselen is an organoselenium drug that inhibits the SARS-CoV-2 main protease (Mpro). Here, the authors co-crystallised Mpro with ebselen and an ebselen derivative and observed an enzyme bound organoselenium covalent adduct in the crystal structures, which was also confirmed by mass spectrometry analysis.
- Kangsa Amporndanai
- , Xiaoli Meng
- & S. Samar Hasnain
-
Article
| Open Access Potent DNA gyrase inhibitors bind asymmetrically to their target using symmetrical bifurcated halogen bonds
Potent DNA gyrase inhibitors bind asymmetrically to their target using symmetrical bifurcated halogen bonds
The mechanism of DNA gyrase inhibitor stabilization of single-strand DNA cleavage breaks by DNA gyrase has been hypothetical. Here, the authors show experimental evidence of the mechanism using a library of inhibitors with improved binding and employ crystal analysis to show bifurcated halogen bonding.
- Anja Kolarič
- , Thomas Germe
- & Marko Anderluh
-
Article
| Open Access A combined high-throughput and high-content platform for unified on-chip synthesis, characterization and biological screening
A combined high-throughput and high-content platform for unified on-chip synthesis, characterization and biological screening
On-chip synthesis and screening has been used to automate drug discovery but on-chip analysis still remains a major limitation. Here, the authors report on a dendrimer-based surface patterning method to create nanodroplet arrays on materials which allow for on-chip high-throughput analysis.
- Maximilian Benz
- , Arndt Asperger
- & Pavel A. Levkin
-
Article
| Open Access Pharmacophore hybridisation and nanoscale assembly to discover self-delivering lysosomotropic new-chemical entities for cancer therapy
Pharmacophore hybridisation and nanoscale assembly to discover self-delivering lysosomotropic new-chemical entities for cancer therapy
Integration of the unique advantages of the fields of drug discovery and drug delivery is invaluable for the advancement of drug development. Here, the authors generate single-drug nanoparticles by hybridising lysomotropic detergents and the bisaminoquinoline-based autophagy inhibitor, and show their therapeutic potential as autophagy-inhibition based combination therapy.
- Zhao Ma
- , Jin Li
- & Yuanpei Li
-
Article
| Open Access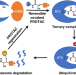 Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry
Enhancing intracellular accumulation and target engagement of PROTACs with reversible covalent chemistry
PROTACs have emerged as promising therapeutic agents but their cellular uptake is often inefficient. Here, the authors show that reversible covalent warhead chemistry improves PROTAC intracellular accumulation and target engagement, and develop a dual inhibitor/degrader of Bruton’s tyrosine kinase
- Wen-Hao Guo
- , Xiaoli Qi
- & Jin Wang
-
Article
| Open Access Cyclic peptide FXII inhibitor provides safe anticoagulation in a thrombosis model and in artificial lungs
Cyclic peptide FXII inhibitor provides safe anticoagulation in a thrombosis model and in artificial lungs
Inhibiting thrombosis without inducing bleeding is a major challenge for anticoagulant agents. Here the authors describe a synthetic FXIIa inhibitor able to efficiently prevent thrombosis in mice and suppress coagulation in artificial lungs in rabbits without increasing the risk of bleeding.
- Jonas Wilbs
- , Xu-Dong Kong
- & Christian Heinis
-
Article
| Open Access Mode of action of teixobactins in cellular membranes
Mode of action of teixobactins in cellular membranes
The natural antibiotic teixobactin kills bacteria by direct binding to their cognate cell wall precursors (Lipid II and III). Here authors use solid-state NMR to reveal the native binding mode of teixobactins and show that teixobactins only weakly bind to Lipid II in anionic cellular membranes.
- Rhythm Shukla
- , João Medeiros-Silva
- & Markus Weingarth
-
Article
| Open Access Selective C-H trifluoromethoxylation of (hetero)arenes as limiting reagent
Selective C-H trifluoromethoxylation of (hetero)arenes as limiting reagent
Selective C-H trifluoromethoxylation of pyridines remains a formidable synthetic challenge. Here, the authors report a silver-mediated late-stage C-H trifluoromethoxylation of arenes and heteroarenes as limiting reagents with trifluoromethoxide anion.
- Zhijie Deng
- , Mingxin Zhao
- & Pingping Tang
-
Article
| Open Access A modular biomimetic strategy for the synthesis of macrolide P-glycoprotein inhibitors via Rh-catalyzed C-H activation
A modular biomimetic strategy for the synthesis of macrolide P-glycoprotein inhibitors via Rh-catalyzed C-H activation
One strategy to address multidrug resistance in cancer is the development of modular methods to access bioactive scaffolds. Here, the authors report a Rh(III)-catalyzed carboxylic acid-directed C(sp2)−H allylation and apply it to the modular synthesis of (Z)-allylic macrolides which enhance antitumoral drug activity.
- Lu Chen
- , Haitian Quan
- & Weibo Yang
-
Article
| Open Access Small molecules that inhibit TNF signalling by stabilising an asymmetric form of the trimer
Small molecules that inhibit TNF signalling by stabilising an asymmetric form of the trimer
While biologics have been successfully applied in TNF antagonist treatments, there are no clinically approved small molecules that target TNF. Here, the authors discover potent small molecule inhibitors of TNF, elucidate their molecular mechanism, and demonstrate TNF inhibition in vitro and in vivo.
- James O’Connell
- , John Porter
- & Alastair Lawson
-
Article
| Open Access Functionalized azetidines via visible light-enabled aza Paternò-Büchi reactions
Functionalized azetidines via visible light-enabled aza Paternò-Büchi reactions
The Aza Paternò-Büchi reaction is arguably among the most direct approaches to functionalized azetidines, which are common medicinal scaffolds. Here, the authors report a mild and selective visible light-enabled intramolecular aza Paternò-Büchi reaction yielding bicyclic azetidines in high yields and diastereoselectivity.
- Marc R. Becker
- , Alistair D. Richardson
- & Corinna S. Schindler
-
Article
| Open Access Gram-scale total synthesis of teixobactin promoting binding mode study and discovery of more potent antibiotics
Gram-scale total synthesis of teixobactin promoting binding mode study and discovery of more potent antibiotics
The presence of the unnatural amino acid l-allo-enduracidine in the cyclic scaffold of teixobactin complicates its total synthesis. Here, the authors developed a convergent strategy for the scalable synthesis teixobactin and found two potent analogous.
- Yu Zong
- , Fang Fang
- & Yu Rao
-
Article
| Open Access Development of a high-throughput strategy for discovery of potent analogues of antibiotic lysocin E
Development of a high-throughput strategy for discovery of potent analogues of antibiotic lysocin E
The depsipeptide Lysocin E has antibacterial activity against methicillin-resistant Staphylococcus aureus. Here, the authors developed a high-throughput one-bead-on-compound method for the synthesis and screening lysocin E derivatives, with several hits being more active than the parent compound.
- Hiroaki Itoh
- , Kotaro Tokumoto
- & Masayuki Inoue
-
Article
| Open Access Catalytic asymmetric acetalization of carboxylic acids for access to chiral phthalidyl ester prodrugs
Catalytic asymmetric acetalization of carboxylic acids for access to chiral phthalidyl ester prodrugs
Phthalidyl esters, commonly used prodrug moieties, are mostly prepared as a racemate. Here, the authors report an N-heterocylcic carbene-catalysed enantioselective acetalization of carboxylic acids and dialdehydes to give phthalidyl esters in high yields with good enantioselectivity.
- Yingguo Liu
- , Qiao Chen
- & Yonggui Robin Chi
-
Article
| Open Access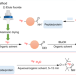 Rapid one-step 18F-radiolabeling of biomolecules in aqueous media by organophosphine fluoride acceptors
Rapid one-step 18F-radiolabeling of biomolecules in aqueous media by organophosphine fluoride acceptors
The synthesis of 18F-labeled positron emission tomography (PET) tracers is difficult and typically requires anhydrous conditions. Here, the authors developed organophosphine precursors that allow for quick, high-yield synthesis of 18F-labeled probes in either organic solvents or aqueous media.
- Huawei Hong
- , Lei Zhang
- & Zijing Li
-
Article
| Open Access Peptide-oligourea hybrids analogue of GLP-1 with improved action in vivo
Peptide-oligourea hybrids analogue of GLP-1 with improved action in vivo
The peptide hormone GLP-1 has the potential to be a remedy for diabetes type II, yet is unstable. Here, the authors synthesized α-peptide-oligourea hybrid analogues of GLP-1 some of which showing significantly prolonged activity in vivo.
- Juliette Fremaux
- , Claire Venin
- & Sébastien R. Goudreau
-
Article
| Open Access The transcription factor STAT5 catalyzes Mannich ligation reactions yielding inhibitors of leukemic cell proliferation
The transcription factor STAT5 catalyzes Mannich ligation reactions yielding inhibitors of leukemic cell proliferation
The oncogene STAT5 is involved in cancer cell proliferation. Here, the authors use STAT5 protein to assemble its own small molecule inhibitors via Mannich ligation (three-component-reactions) and show that the resultant ligands can inhibit the proliferation of cancer cells in a mouse model.
- Ee Lin Wong
- , Eric Nawrotzky
- & Jörg Rademann
-
Article
| Open Access Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia
Discovery of Mcl-1-specific inhibitor AZD5991 and preclinical activity in multiple myeloma and acute myeloid leukemia
High expression of Mcl-1 promotes tumorigenesis and resistance to anticancer therapies. Here they report a macrocyclic molecule with high selectivity and affinity for Mcl-1 that exhibits potent anti-tumor effects as single agent and in combination with bortezomib or venetoclax in preclinical models of multiple myeloma and acute myeloid leukemia.
- Adriana E. Tron
- , Matthew A. Belmonte
- & Alexander W. Hird
-
Article
| Open Access A therapeutic approach to pantothenate kinase associated neurodegeneration
A therapeutic approach to pantothenate kinase associated neurodegeneration
Mutations in pantotenate kinase (PANK) cause neurodegneration. Here the authors carry out achemical screen and identify a PANK activator that is orally available, crosses the blood brain barrierand show that it effecttive in improving pathology and life span in a mouse model of the disease.
- Lalit Kumar Sharma
- , Chitra Subramanian
- & Suzanne Jackowski
-
Article
| Open Access Clopidogrel as a donor probe and thioenol derivatives as flexible promoieties for enabling H2S biomedicine
Clopidogrel as a donor probe and thioenol derivatives as flexible promoieties for enabling H2S biomedicine
Hydrogen sulphide (H2S) is a gaseous signalling molecule, which has shown therapeutic value. Here, the authors show that a thioenol metabolite of the antithrombotic drug clopidogrel is an efficient H2S donor and masked thioenols can be linked to existing compounds to develop H2S-releasing agents.
- Yaoqiu Zhu
- , Elkin L. Romero
- & Bin Geng
-
Article
| Open Access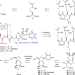 Total synthesis and antimicrobial evaluation of natural albomycins against clinical pathogens
Total synthesis and antimicrobial evaluation of natural albomycins against clinical pathogens
Albomycins are promising drug candidates for the treatment of bacterial infections. Here, the authors describe the total syntheses of albomycins δ1, δ2, and ε, and evaluate their antimicrobial activity, identifying albomycin δ2 as a strong agent against S. pneumoniae and S. aureus infections.
- Zihua Lin
- , Xiaobo Xu
- & Yun He
-
Article
| Open Access Scalable synthesis enabling multilevel bio-evaluations of natural products for discovery of lead compounds
Scalable synthesis enabling multilevel bio-evaluations of natural products for discovery of lead compounds
Isodon diterpenoids, promising anti-cancer agents found in certain tropical plants, are difficult to obtain. Here, the authors developed a synthetic strategy to synthesise several different members of this group, including neolaxiflorin L which emerged from this study as a promising drug candidate.
- Lizhi Zhu
- , Wenjing Ma
- & Chi-Sing Lee
-
Article
| Open Access A small molecule inhibitor of Rheb selectively targets mTORC1 signaling
A small molecule inhibitor of Rheb selectively targets mTORC1 signaling
Aberrant mTORC1 signaling is linked to several chronic diseases. Here, the authors develop a small molecule inhibitor that binds the small G-protein Rheb and selectively blocks mTORC1 signaling, holding potential for therapeutic applications.
- Sarah J. Mahoney
- , Sridhar Narayan
- & Eddine Saiah
-
Article
| Open Access Targeted NUDT5 inhibitors block hormone signaling in breast cancer cells
Targeted NUDT5 inhibitors block hormone signaling in breast cancer cells
NUDIX hydrolases are an important family of nucleotide-metabolizing enzymes. Here, the authors identify potent, small molecule inhibitors of NUDT5, which is implicated in ADP-ribose and 8-oxo-guanine metabolism, and confirm its role in gene regulation and proliferation in breast cancer cells.
- Brent D. G. Page
- , Nicholas C. K. Valerie
- & Thomas Helleday
-
Article
| Open Access Protein-inspired antibiotics active against vancomycin- and daptomycin-resistant bacteria
Protein-inspired antibiotics active against vancomycin- and daptomycin-resistant bacteria
The antibiotic vancomycin inhibits bacterial cell wall synthesis by binding to a membrane-associated precursor. Here, Blaskovich et al. synthesize vancomycin derivatives containing lipophilic peptide moieties that enhance membrane affinity and in vivo activities against glycopeptide-resistant strains.
- Mark A. T. Blaskovich
- , Karl A. Hansford
- & Matthew A. Cooper
-
Article
| Open Access Diastereo- and enantioselective [3 + 3] cycloaddition of spirocyclopropyl oxindoles using both aldonitrones and ketonitrones
Diastereo- and enantioselective [3 + 3] cycloaddition of spirocyclopropyl oxindoles using both aldonitrones and ketonitrones
Chiral spirocyclic compounds are important structural motifs for drug discovery. Here, the authors report a synthetic route to spirocycles based on enantioselective cycloaddition of activated spirocyclopropyl oxindoles, which act as donor-acceptor cyclopropanes.
- Peng-Wei Xu
- , Jia-Kuan Liu
- & Jian Zhou
-
Article
| Open Access Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors
Assay interference and off-target liabilities of reported histone acetyltransferase inhibitors
A substantial obstacle in basic research is the use of poorly validated tool compounds with purported useful biological functions. Here, the authors systematically profile widely used histone acetyltransferase inhibitors and find that the majority are nonselective interference compounds.
- Jayme L. Dahlin
- , Kathryn M. Nelson
- & Michael A. Walters
-
Article
| Open Access Organocatalytic enantio- and diastereoselective cycloetherification via dynamic kinetic resolution of chiral cyanohydrins
Organocatalytic enantio- and diastereoselective cycloetherification via dynamic kinetic resolution of chiral cyanohydrins
Enantioselective synthesis of six-membered oxacycles with multiple stereogenic centres is essential for the discovery of new therapeutic agents. Here the authors show organocatalytic cycloetherification for the highly enantio- and diastereoselective synthesis of tetrahydropyrans.
- Naoki Yoneda
- , Yuki Fujii
- & Seijiro Matsubara
-
Article
| Open Access A potent series targeting the malarial cGMP-dependent protein kinase clears infection and blocks transmission
A potent series targeting the malarial cGMP-dependent protein kinase clears infection and blocks transmission
Protein kinases are promising drug targets for treatment of malaria. Here, starting with a medicinal chemistry approach, Baker et al. generate an imidazopyridine that selectively targets Plasmodium falciparum PKG, inhibits blood stage parasite growth in vitro and in mice and blocks transmission to mosquitoes.
- David A. Baker
- , Lindsay B. Stewart
- & Simon A. Osborne
-
Article
| Open Access Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib
Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib
Lorlatinib—a ROS1/ALK inhibitor—is currently undergoing clinical trials for the treatment of non-small cell lung cancers. Here the authors develop synthetic routes to11C- and 18F-labelled lorlatinib, with subsequent PET imaging showing good blood brain barrier permeability in non-human primates.
- Thomas Lee Collier
- , Marc D. Normandin
- & Neil Vasdev
-
Article
| Open Access Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies
Discovery of first-in-class reversible dual small molecule inhibitors against G9a and DNMTs in hematological malignancies
Epigenetic drugs are emerging as a powerful therapeutic option for cancer treatment. Here, the authors synthesized selective chemical probes that simultaneously inhibit the G9a and DNMTs methyltransferase activity and demonstrate their anti-tumour activity usingin vitro and in vivomodels of haematological neoplasia.
- Edurne San José-Enériz
- , Xabier Agirre
- & Felipe Prosper
-
Article
| Open Access A tetraoxane-based antimalarial drug candidate that overcomes PfK13-C580Y dependent artemisinin resistance
A tetraoxane-based antimalarial drug candidate that overcomes PfK13-C580Y dependent artemisinin resistance
Artemisinin-resistantPlasmodium is an increasing problem. Here, using a medicinal chemistry programme, the authors identify a tetraoxane-based drug candidate that shows no cross-resistance with an artemisinin-resistant strain (PfK13-C580Y) and is efficient in Plasmodiummouse models.
- Paul M. O’Neill
- , Richard K. Amewu
- & Stephen A. Ward
-
Article
| Open Access Allosteric cross-talk in chromatin can mediate drug-drug synergy
Allosteric cross-talk in chromatin can mediate drug-drug synergy
Allostery and drug-drug synergism can yield potential novel therapies with existing molecules. Here, the authors provide evidence that two unrelated drugs have increased cytotoxicity in cancer cells, which coincides with increased formation of chromatin adducts.
- Zenita Adhireksan
- , Giulia Palermo
- & Curt A. Davey
-
Article
| Open Access Inhibition of delta-secretase improves cognitive functions in mouse models of Alzheimer’s disease
Inhibition of delta-secretase improves cognitive functions in mouse models of Alzheimer’s disease
Delta-secretases are associated with Alzheimer’s disease (AD) as they cleave both amyloid precursor protein and tau. Here the authors develop a series of orally bioactive small molecule delta-secretase inhibitors and report its therapeutic effects in mouse models of AD.
- Zhentao Zhang
- , Obiamaka Obianyo
- & Keqiang Ye
-
Article
| Open Access A chemical chaperone improves muscle function in mice with a RyR1 mutation
A chemical chaperone improves muscle function in mice with a RyR1 mutation
Mutations in the RyR1 channel cause core myopathies. Here the authors show that ER stress and the unfolded protein response underlie the pathology caused by a common RyR1 channel mutation, and show that treatment with a chemical chaperone restores muscle function in mice.
- Chang Seok Lee
- , Amy D. Hanna
- & Susan L. Hamilton
-
Article
| Open Access Sansanmycin natural product analogues as potent and selective anti-mycobacterials that inhibit lipid I biosynthesis
Sansanmycin natural product analogues as potent and selective anti-mycobacterials that inhibit lipid I biosynthesis
Drug resistant tuberculosis (TB) infections are emerging at a high rate, with only few therapeutic options currently available. Here, the authors report synthetic analogues of the natural product sansanmycin that target mycobacterial cell wall biosynthesis and represent potent leads for improved anti-TB treatments.
- Anh T. Tran
- , Emma E. Watson
- & Richard J. Payne
-
Article
| Open Access In situ click chemistry generation of cyclooxygenase-2 inhibitors
In situ click chemistry generation of cyclooxygenase-2 inhibitors
Traditional inflammation and pain relief drugs target both cyclooxygenase 1 and 2 (COX-1 and COX-2), causing severe side effects. Here, the authors use in situ click chemistry to develop COX-2 specific inhibitors with high in vivo anti-inflammatory activity.
- Atul Bhardwaj
- , Jatinder Kaur
- & Frank Wuest
-
Article
| Open Access Arylmethylamino steroids as antiparasitic agents
Arylmethylamino steroids as antiparasitic agents
Steroid units can facilitate membrane permeation and bioavailability in drugs. Here, using a medicinal chemistry program, Krieget al. identify an arylmethylamino steroid that kills Plasmodium parasites, likely through a chelate-based quinone methide mechanism, and has activity against Schistosoma mansoni.
- Reimar Krieg
- , Esther Jortzik
- & Katja Becker
-
Article
| Open Access Total synthesis of feglymycin based on a linear/convergent hybrid approach using micro-flow amide bond formation
Total synthesis of feglymycin based on a linear/convergent hybrid approach using micro-flow amide bond formation
Feglymycin is a biologically active peptide but a challenging synthetic target due to the highly racemizable nature of the 3,5-dihydroxyphenylglycine groups. Here the authors report the synthesis of feglymycin using a microflow system, allowing amide bond formation without severe racemization.
- Shinichiro Fuse
- , Yuto Mifune
- & Hiroshi Tanaka
-
Article
| Open Access Mechanistic evaluation and transcriptional signature of a glutathione S-transferase omega 1 inhibitor
Mechanistic evaluation and transcriptional signature of a glutathione S-transferase omega 1 inhibitor
Glutathione S-transferase omega 1 (GSTO1) is an atypical GST isoform overexpressed in several cancers that has been implicated in drug resistance. Here the authors identify a small molecule inhibitor of GSTO1 that effectively inhibits tumor growth in colon cancer models, and establish its mechanism of action.
- Kavya Ramkumar
- , Soma Samanta
- & Nouri Neamati
-
Article
| Open Access Irreversible inhibitors of the 3C protease of Coxsackie virus through templated assembly of protein-binding fragments
Irreversible inhibitors of the 3C protease of Coxsackie virus through templated assembly of protein-binding fragments
Molecular fragments are useful tools in drug-discovery but they might be hard to identify due to their weak affinity to the targets. Here, the authors use a protein-templated assembly to design high affinity inhibitors of Coxsackie virus 3C protease, a pharmacological target against enteroviral infections.
- Daniel Becker
- , Zuzanna Kaczmarska
- & Jörg Rademann
-
Article
| Open Access Structure-guided development of heterodimer-selective GPCR ligands
Structure-guided development of heterodimer-selective GPCR ligands
G protein-coupled receptors (GPCRs) are involved in key signalling pathways and represent important targets for the treatment of neurological and psychiatric disorders. Here, the authors describe powerful bivalent ligands that efficiently bind to therapeutically relevant GPCR heterodimers
- Harald Hübner
- , Tamara Schellhorn
- & Peter Gmeiner
-
Article
| Open Access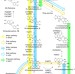 Engineering biosynthesis of the anticancer alkaloid noscapine in yeast
Engineering biosynthesis of the anticancer alkaloid noscapine in yeast
Noscapine is a potential anticancer drug that is traditionally isolated from the opium poppy Papaver somniferum. Here, Li and Smolke reconstitute the noscapine gene cluster in Saccharomyces cerevisiae, to achieve the microbial production of noscapine and related pathway intermediates, and provide new insights into the biosynthesis of noscapine.
- Yanran Li
- & Christina D. Smolke
-
Article
| Open Access Drug design from the cryptic inhibitor envelope
Drug design from the cryptic inhibitor envelope
The conformational dynamics of a compound has a large effect on ligand/receptor binding. Here, the authors employ NMR spectroscopy to study ligand binding to the enzyme LpxC, discovering an inhibitor envelope that was not identifiable by crystallography and subsequently developing a highly potent inhibitor.
- Chul-Jin Lee
- , Xiaofei Liang
- & Pei Zhou
-
Article
| Open Access Development of high-yield influenza A virus vaccine viruses
Development of high-yield influenza A virus vaccine viruses
The availability of high-yield virus strains remains an important bottleneck in the rapid production of influenza vaccines. Here, the authors report the development of influenza A vaccine backbone that improves the virus yield of various seasonal and pandemic influenza vaccine strains in cell culture.
- Jihui Ping
- , Tiago J.S. Lopes
- & Yoshihiro Kawaoka
-
Article
| Open Access The key role of the scaffold on the efficiency of dendrimer nanodrugs
The key role of the scaffold on the efficiency of dendrimer nanodrugs
The biological properties of dendrimers are thought to be largely dependent on the chemical nature of their surface. Here, the authors show that the internal scaffold of dendritic nanodrugs strongly influences their bioactivity, based on convergent information from biology and computation results.
- Anne-Marie Caminade
- , Séverine Fruchon
- & Rémy Poupot
-
Article
| Open Access Caenorhabditis elegans is a useful model for anthelmintic discovery
Caenorhabditis elegans is a useful model for anthelmintic discovery
Screening for new anthelmintic compounds that are active against parasitic nematodes is costly and labour intensive. Here, the authors use the non-parasitic nematode Caenorhabditis elegansto identify 30 anthelmintic lead compounds in an effective and cost-efficient manner.
- Andrew R. Burns
- , Genna M. Luciani
- & Peter J. Roy
-
Article
| Open Access Transfer hydrogenation catalysis in cells as a new approach to anticancer drug design
Transfer hydrogenation catalysis in cells as a new approach to anticancer drug design
Organometallic complexes are effective hydrogenation catalysts for organic reactions. Here the authors report for the first time that transfer hydrogenation catalysis can take place inside the cell and could be used as a novel anticancer strategy.
- Joan J. Soldevila-Barreda
- , Isolda Romero-Canelón
- & Peter J. Sadler
-
Article
| Open Access Ligand substitutions between ruthenium–cymene compounds can control protein versus DNA targeting and anticancer activity
Ligand substitutions between ruthenium–cymene compounds can control protein versus DNA targeting and anticancer activity
Ruthenium-cymene-based compounds are investigated as potential anticancer drugs. Here, Adhireksan et al.study two ruthenium-containing compounds with varying cytotoxicity and show that differences in ligand structure may explain their activity and binding to different subcellular targets.
- Zenita Adhireksan
- , Gabriela E. Davey
- & Curt A. Davey
 Exploring protein hotspots by optimized fragment pharmacophores
Exploring protein hotspots by optimized fragment pharmacophores