
Featured
-
-
Letter |
 TP53 loss creates therapeutic vulnerability in colorectal cancer
TP53 loss creates therapeutic vulnerability in colorectal cancer
Genomic deletion of the tumour suppressor TP53 frequently includes other neighbouring genes, such as the POLR2A housekeeping gene that encodes a crucial RNA polymerase II subunit; suppression of POLR2A with α-amanitin or by RNA interference selectively inhibits the tumorigenic potential of cancer cells, and in mouse models of cancer, tumours can be selectively targeted with α-amanitin coupled to antibodies, suggesting new therapeutic approaches for human cancers.
- Yunhua Liu
- , Xinna Zhang
- & Xiongbin Lu
-
Spotlight |
 Spotlight on Cancer Research
Spotlight on Cancer Research
-
Letter |
 The evolutionary history of lethal metastatic prostate cancer
The evolutionary history of lethal metastatic prostate cancer
The subclonal composition of human prostate tumours and their metastases has been mapped by whole-genome sequencing, thus establishing the evolutionary trees behind the development and spread of these cancers; an important observation was that metastases could be re-seeded multiple times, and spread from one tumour to another was frequently seen.
- Gunes Gundem
- , Peter Van Loo
- & G. Steven Bova
-
Letter |
 Differential DNA mismatch repair underlies mutation rate variation across the human genome
Differential DNA mismatch repair underlies mutation rate variation across the human genome
An analysis of how regional mutation rates vary across 652 tumours identifies variable DNA mismatch repair as the basis of the characteristic regional variation in mutation rates seen across the human genome; the results show that differential DNA repair, rather than differential mutation supply, is likely to be the primary cause of this variation.
- Fran Supek
- & Ben Lehner
-
Letter
| Open Access Cell-of-origin chromatin organization shapes the mutational landscape of cancer
Cell-of-origin chromatin organization shapes the mutational landscape of cancer
An analysis of cell-type-specific epigenomic features reveals a relationship between epigenomic and mutational profiles; chromatin characteristics can explain a large proportion of mutational variance in cancer genomes and the mutational distribution can identify the probable cell type from which a given cancer originated from.
- Paz Polak
- , Rosa Karlić
- & Shamil R. Sunyaev
-
Letter |
 Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution
Dynamics of genomic clones in breast cancer patient xenografts at single-cell resolution
Deep-genome and single-cell sequencing analyses of patient-derived breast cancer xenografts reveal extensive, dynamic and reproducible changes in intra-tumoral mutational clonal composition on engraftment and serial propagation.
- Peter Eirew
- , Adi Steif
- & Samuel Aparicio
-
Letter |
 The mutational landscapes of genetic and chemical models of Kras-driven lung cancer
The mutational landscapes of genetic and chemical models of Kras-driven lung cancer
Whole-exome sequencing is used to compare the mutational landscape of adenomas from three mouse models of non-small-cell lung cancer, induced either by exposure to carcinogens or by genetic mutation of Kras; the results reveal that the two types of tumour have different mutational profiles and adopt different routes to tumour development.
- Peter M. K. Westcott
- , Kyle D. Halliwill
- & Allan Balmain
-
Article |
 Clonal evolution in breast cancer revealed by single nucleus genome sequencing
Clonal evolution in breast cancer revealed by single nucleus genome sequencing
To investigate genomic diversity within tumours, a new type of whole-genome and exome single cell sequencing has been developed using G2/M nuclei; the technique was used to sequence single nuclei from an oestrogen-positive breast cancer and a triple-negative ductal carcinoma—aneuploidy rearrangements emerged as early events in tumour formation and then point mutations evolved gradually over time.
- Yong Wang
- , Jill Waters
- & Nicholas E. Navin
-
Letter |
 Putative cis-regulatory drivers in colorectal cancer
Putative cis-regulatory drivers in colorectal cancer
Examination of allele-specific expression identifies 71 genes with excess somatic cis-regulatory effects in colorectal cancer (CRC), and 1,693 and 948 expression quantitative trait loci (eQTLs) in normal samples and tumours, respectively (with 36% of tumour eQTLs exclusive to CRC); tumour-specific eQTLs are more enriched for low CRC genome-wide association study P values and accumulate more somatic mutations than shared eQTLs, suggesting a role as germline-derived cancer regulatory drivers.
- Halit Ongen
- , Claus L. Andersen
- & Emmanouil T. Dermitzakis
-
Article
| Open Access Comprehensive molecular characterization of gastric adenocarcinoma
Comprehensive molecular characterization of gastric adenocarcinoma
The Cancer Genome Atlas reports on molecular evaluation of 295 primary gastric adenocarcinomas and proposes a new classification of gastric cancers into 4 subtypes, which should help with clinical assessment and trials of targeted therapies.
- Adam J. Bass
- , Vesteinn Thorsson
- & Jia Liu
-
Article |
 Proteogenomic characterization of human colon and rectal cancer
Proteogenomic characterization of human colon and rectal cancer
Proteome analysis of The Cancer Genome Atlas (TCGA) colorectal cancer specimens reveals that DNA- or RNA-level measurements cannot reliably predict protein abundance, colorectal tumours can be separated into distinct proteotypes, and that copy number alterations drive mRNA abundance changes but few extend to protein-level changes.
- Bing Zhang
- , Jing Wang
- & R. Reid Townsend
-
Letter |
 Metastasis-suppressor transcript destabilization through TARBP2 binding of mRNA hairpins
Metastasis-suppressor transcript destabilization through TARBP2 binding of mRNA hairpins
Linear sequence elements within messenger RNAs are known to be targeted by regulatory factors such as microRNAs for degradation, a process that has been implicated in disease; now, non-linear regulatory structural elements within mRNAs are shown also to be targeted, with the resulting mRNA destabilization mediating breast cancer metastasis.
- Hani Goodarzi
- , Steven Zhang
- & Sohail F. Tavazoie
-
Article
| Open Access Comprehensive molecular profiling of lung adenocarcinoma
Comprehensive molecular profiling of lung adenocarcinoma
An integrated transcriptome, genome, methylome and proteome analysis of over 200 lung adenocarcinomas reveals high rates of somatic mutations, 18 statistically significantly mutated genes including RIT1 and MGA, splicing changes, and alterations in MAPK and PI(3)K pathway activity.
- Eric A. Collisson
- , Joshua D. Campbell
- & Ming-Sound Tsao
-
Article |
 Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma
Enhancer hijacking activates GFI1 family oncogenes in medulloblastoma
Focusing on two ill-characterized subtypes of medulloblastoma (group 3 and group 4), this study identifies prevalent genomic structural variants that are restricted to these two subtypes and independently bring together coding regions of GFI1 family proto-oncogenes with active enhancer elements, leading to their mutually exclusive oncogenic activation.
- Paul A. Northcott
- , Catherine Lee
- & Stefan M. Pfister
-
Letter |
 PVT1 dependence in cancer with MYC copy-number increase
PVT1 dependence in cancer with MYC copy-number increase
Pvt1 overexpression in mice contributes to high Myc levels due to 8q24.21 gain and to MYC-driven tumorigenesis.
- Yuen-Yi Tseng
- , Branden S. Moriarity
- & Anindya Bagchi
-
Letter |
 Novel somatic and germline mutations in intracranial germ cell tumours
Novel somatic and germline mutations in intracranial germ cell tumours
Intracranial germ cell tumours are rare tumours affecting mainly male adolescents, mainly in Asia; here the authors identify frequent mutations in the KIT/RAS and AKT/mTOR signalling pathways as well as rare germline variants in JMJD1C, suggesting potential therapeutic strategies focusing on the inhibition of KIT/RAS activation and the AKT1/mTOR pathway.
- Linghua Wang
- , Shigeru Yamaguchi
- & Ching C. Lau
-
Letter |
 Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing
Decoding the regulatory landscape of medulloblastoma using DNA methylation sequencing
Medulloblastoma is a malignant childhood brain tumour presenting major clinical challenges; here, a comprehensive genome-wide DNA methylation data set from human and mouse tumours, coupled with analysis of histone modifications, RNA transcripts and genome sequencing, uncovers a wealth of alterations that provide insights into the epigenetic regulation of transcription and genome organization in medulloblastoma pathogenesis.
- Volker Hovestadt
- , David T. W. Jones
- & Peter Lichter
-
Letter |
 Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia
Constitutional and somatic rearrangement of chromosome 21 in acute lymphoblastic leukaemia
A rare constitutional translocation between chromosomes 15 and 21 predisposes to catastrophic chromosomal damage followed by amplification of megabase regions, causing a specific subtype of acute lymphoblastic leukaemia.
- Yilong Li
- , Claire Schwab
- & Christine J. Harrison
-
Letter |
 Identification of genomic alterations in oesophageal squamous cell cancer
Identification of genomic alterations in oesophageal squamous cell cancer
Using whole-genome and whole-exome sequencing, this study identifies eight significantly mutated genes in oesophageal squamous cell cancer, including two genes, ADAM29 and FAM135B, not previously associated with this cancer type.
- Yongmei Song
- , Lin Li
- & Qimin Zhan
-
Article
| Open Access Comprehensive molecular characterization of urothelial bladder carcinoma
Comprehensive molecular characterization of urothelial bladder carcinoma
This paper reports integrative molecular analyses of urothelial bladder carcinoma at the DNA, RNA, and protein levels performed as part of The Cancer Genome Atlas project; recurrent mutations were found in 32 genes, including those involved in cell-cycle regulation, chromatin regulation and kinase signalling pathways; chromatin regulatory genes were more frequently mutated in urothelial carcinoma than in any other common cancer studied so far.
- John N. Weinstein
- , Rehan Akbani
- & Greg Eley
-
Article |
 Discovery and saturation analysis of cancer genes across 21 tumour types
Discovery and saturation analysis of cancer genes across 21 tumour types
Large-scale genomic analysis of somatic point mutations in exomes from tumour–normal pairs across 21 cancer types identifies most known cancer genes in these tumour types as well as 33 genes not known to be significantly mutated, and down-sampling analysis indicates that larger sample sizes will reveal many more genes mutated at clinically important frequencies.
- Michael S. Lawrence
- , Petar Stojanov
- & Gad Getz
-
Letter |
 Landscape of genomic alterations in cervical carcinomas
Landscape of genomic alterations in cervical carcinomas
Whole-exome sequencing and analysis of 115 cervical carcinoma–normal paired samples, in addition to transcriptome and whole-genome sequencing for a subset of these tumours, reveal novel genes mutated at significant levels within this cohort and provide evidence that HPV integration is a common mechanism for target gene overexpression; results also compare mutational landscapes between squamous cell carcinomas and adenocarcinomas.
- Akinyemi I. Ojesina
- , Lee Lichtenstein
- & Matthew Meyerson
-
Analysis |
 Inconsistency in large pharmacogenomic studies
Inconsistency in large pharmacogenomic studies
This Analysis compares two large-scale pharmacogenomic data sets that catalogued the sensitivity of a large number of cancer cell lines to approved and potential drugs, and finds that whereas the gene expression data are largely concordant between the two studies, the reported drug sensitivity measures and subsequently their association with genomic features are highly discordant.
- Benjamin Haibe-Kains
- , Nehme El-Hachem
- & John Quackenbush
-
Article
| Open Access Mutational landscape and significance across 12 major cancer types
Mutational landscape and significance across 12 major cancer types
As part of The Cancer Genome Atlas Pan-Cancer effort, data analysis for point mutations and small indels from 3,281 tumours and 12 tumour types is presented; among the findings are 127 significantly mutated genes from cellular processes with both established and emerging links in cancer, and an indication that the number of driver mutations required for oncogenesis is relatively small.
- Cyriac Kandoth
- , Michael D. McLellan
- & Li Ding
-
Letter |
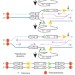 Two replication fork maintenance pathways fuse inverted repeats to rearrange chromosomes
Two replication fork maintenance pathways fuse inverted repeats to rearrange chromosomes
Stalling of replication forks in sequences that have non-allelic repeats can lead to genomic rearrangements; here two pathways consistent with homologous recombination and error-free post-replication repair fuse identical and mismatched repeats, respectively, thus inducing chromosomal rearrangements in mouse embryonic stem cells.
- Lingchuan Hu
- , Tae Moon Kim
- & Paul Hasty
-
Article |
 RNAi screens in mice identify physiological regulators of oncogenic growth
RNAi screens in mice identify physiological regulators of oncogenic growth
Here, the first genome-wide in vivo RNA interference screens in a mammalian animal model are reported: genes involved in normal and abnormal epithelial cell growth are studied in developing skin tissue in mouse embryos, and among the findings, β-catenin is shown to act as an antagonist to normal epithelial cell growth as well as promoting oncogene-driven growth.
- Slobodan Beronja
- , Peter Janki
- & Elaine Fuchs
-
Letter |
 The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line
The haplotype-resolved genome and epigenome of the aneuploid HeLa cancer cell line
Haplotype-resolved whole-genome sequencing of the HeLa CCL-2 strain shows that HeLa is relatively stable in terms of point variation; integration of several data sets reveals strong, haplotype-specific activation of the proto-oncogene MYC by the human papilloma virus type 18 genome, and enables the relationship between gene dosage and expression to be examined.
- Andrew Adey
- , Joshua N. Burton
- & Jay Shendure
-
Outlook |
 Genetics: Written in blood
Genetics: Written in blood
Technologies that rapidly sequence DNA reveal deep genetic diversity both within and among individuals with leukaemia.
- Sarah DeWeerdt
-
Article
| Open Access Comprehensive molecular characterization of clear cell renal cell carcinoma
Comprehensive molecular characterization of clear cell renal cell carcinoma
The Cancer Genome Atlas Research Network reports an integrative analysis of more than 400 samples of clear cell renal cell carcinoma based on genomic, DNA methylation, RNA and proteomic characterisation; frequent mutations were identified in the PI(3)K/AKT pathway, suggesting this pathway might be a potential therapeutic target, among the findings is also a demonstration of metabolic remodelling which correlates with tumour stage and severity.
- Chad J. Creighton
- , Margaret Morgan
- & Heidi J. Sofia.
-
Letter |
 Mutational heterogeneity in cancer and the search for new cancer-associated genes
Mutational heterogeneity in cancer and the search for new cancer-associated genes
As the sample size in cancer genome studies increases, the list of genes identified as significantly mutated is likely to include more false positives; here, this problem is identified as stemming largely from mutation heterogeneity, and a new analytical methodology designed to overcome this problem is described.
- Michael S. Lawrence
- , Petar Stojanov
- & Gad Getz
-
Letter |
 The shaping and functional consequences of the microRNA landscape in breast cancer
The shaping and functional consequences of the microRNA landscape in breast cancer
MicroRNA profiling of 1,302 human breast tumour samples provides an overview of the miRNA landscape and its regulation, revealing context-dependent interactions, broad prognostic value of miRNA signatures and an important modulatory role for miRNAs in the biology of breast tumours devoid of copy-number aberrations.
- Heidi Dvinge
- , Anna Git
- & Carlos Caldas
-
Article
| Open Access Integrated genomic characterization of endometrial carcinoma
Integrated genomic characterization of endometrial carcinoma
An integrative genomic analysis of several hundred endometrial carcinomas shows that a minority of tumour samples carry copy number alterations or TP53 mutations and many contain key cancer-related gene mutations, such as those involved in canonical pathways and chromatin remodelling; a reclassification of endometrial tumours into four distinct types is proposed, which may have an effect on patient treatment regimes.
- Douglas A. Levine
- , Gad Getz
- & Douglas A. Levine
-
Letter |
 Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA
Non-invasive analysis of acquired resistance to cancer therapy by sequencing of plasma DNA
A proof of principle study shows that by exome sequencing of cell-free circulating DNA from cancer patient plasma samples, the genomic evolution of metastatic cancers and the acquisition of resistance in response to therapy can be tracked over time.
- Muhammed Murtaza
- , Sarah-Jane Dawson
- & Nitzan Rosenfeld
-
Letter |
 Replication stress links structural and numerical cancer chromosomal instability
Replication stress links structural and numerical cancer chromosomal instability
A mechanism to explain chromosomal instability (CIN) in colorectal cancer is demonstrated; three new CIN-suppressor genes (PIGN, MEX3C and ZNF516) encoded on chromosome 18q are identified, the loss of which leads to DNA replication stress, resulting in structural and numerical chromosome segregation errors, which are shown to be identical to phenotypes seen in CIN cells.
- Rebecca A. Burrell
- , Sarah E. McClelland
- & Charles Swanton
-
News Feature |
 Genomics: The single life
Genomics: The single life
Sequencing DNA from individual cells is changing the way that researchers think of humans as a whole.
- Brian Owens
-
Article |
 Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes
Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes
Exome sequencing and copy number analysis are used to define genomic aberrations in early sporadic pancreatic ductal adenocarcinoma; among the findings are mutations in genes involved in chromatin modification and DNA damage repair, and frequent and diverse somatic aberrations in genes known as embryonic regulators of axon guidance.
- Andrew V. Biankin
- , Nicola Waddell
- & Sean M. Grimmond
-
News |
 Studies offer ‘panoramic view’ of lung cancer
Studies offer ‘panoramic view’ of lung cancer
Three genome-sequencing trials may help to revamp treatments for the world’s most deadly cancer.
- Monya Baker
-
Article
| Open Access Comprehensive genomic characterization of squamous cell lung cancers
Comprehensive genomic characterization of squamous cell lung cancers
Comprehensive analyses of 178 lung squamous cell carcinomas by The Cancer Genome Atlas project show that the tumour type is characterized by complex genomic alterations, with statistically recurrent mutations in 11 genes, including TP53 in nearly all samples; a potential therapeutic target is identified in most of the samples studied.
- Peter S. Hammerman
- , Michael S. Lawrence
- & Matthew Meyerson
-
Letter
| Open Access Recurrent R-spondin fusions in colon cancer
Recurrent R-spondin fusions in colon cancer
Exomes, transcriptomes and copy-number alterations in a sample of more than 70 primary human colonic tumours were analysed in an attempt to characterize the genomic landscape; in addition to finding alterations in genes associated with commonly mutated signalling pathways, recurrent gene fusions involving R-spondin family members were also found to occur in approximately 10% of colonic tumours, revealing a potential new therapeutic target.
- Somasekar Seshagiri
- , Eric W. Stawiski
- & Frederic J. de Sauvage
-
Letter |
 Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics
Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics
RNA sequencing of Burkitt lymphoma tumours allows identification of mutations affecting the transcription factor TCF3, its negative regulator ID3 and the cell cycle regulator CCND3; these pathways reveal new targets for potential therapeutic intervention.
- Roland Schmitz
- , Ryan M. Young
- & Louis M. Staudt
-
Letter |
 Chromatin organization is a major influence on regional mutation rates in human cancer cells
Chromatin organization is a major influence on regional mutation rates in human cancer cells
Mutation rates in cancer genomes are closely related to chromatin organization, indicating that the arrangement of the genome into heterochromatin- and euchromatin-like domains may be a dominant influence on variation in regional mutation rate in human somatic cells.
- Benjamin Schuster-Böckler
- & Ben Lehner
-
Letter |
 Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins
Interpreting cancer genomes using systematic host network perturbations by tumour virus proteins
Combining analysis of host proteome and transcriptome perturbations induced by tumour virus proteins with ongoing genome-wide studies of cancer facilitates the prioritization of cancer genes.
- Orit Rozenblatt-Rosen
- , Rahul C. Deo
- & Marc Vidal
-
-
Article
| Open Access Novel mutations target distinct subgroups of medulloblastoma
Novel mutations target distinct subgroups of medulloblastoma
Whole-genome sequencing of medulloblastoma samples reveals several recurrent mutations in genes not previously implicated in the disease, many of which affect components of the epigenetic machinery in different disease subgroups.
- Giles Robinson
- , Matthew Parker
- & Richard J. Gilbertson
-
Article
| Open Access Whole-genome analysis informs breast cancer response to aromatase inhibition
Whole-genome analysis informs breast cancer response to aromatase inhibition
Whole-genome analysis of oestrogen-receptor-positive tumours in patients treated with aromatase inhibitors show that distinct phenotypes are associated with specific patterns of somatic mutations; however, most recurrent mutations are relatively infrequent so prospective clinical trials will require comprehensive sequencing and large study populations.
- Matthew J. Ellis
- , Li Ding
- & Elaine R. Mardis
-
Letter |
 A tumour suppressor network relying on the polyamine–hypusine axis
A tumour suppressor network relying on the polyamine–hypusine axis
AMD1 and eIF5A are identified as two genes involved in the polyamine–hypusine pathway, a new tumour suppressor network regulating apoptosis.
- Claudio Scuoppo
- , Cornelius Miething
- & Scott W. Lowe
-
Letter |
 The mutational landscape of lethal castration-resistant prostate cancer
The mutational landscape of lethal castration-resistant prostate cancer
Exome sequencing is used to investigate the role of mutations and copy number aberrations in metastatic castration-resistant prostate cancer, revealing recurrent mutations in multiple chromatin/histone modifying genes, as well as genes involved in androgen signalling.
- Catherine S. Grasso
- , Yi-Mi Wu
- & Scott A. Tomlins
-
Letter |
 The landscape of cancer genes and mutational processes in breast cancer
The landscape of cancer genes and mutational processes in breast cancer
A study of breast cancers shows that the number of somatic mutations in each varies markedly and is strongly correlated with age at diagnosis and cancer histological grade.
- Philip J. Stephens
- , Patrick S. Tarpey
- & Michael R. Stratton
-
Letter
| Open Access Melanoma genome sequencing reveals frequent PREX2 mutations
Melanoma genome sequencing reveals frequent PREX2 mutations
Whole-genome sequencing of 25 metastatic melanomas and matched germline DNA in humans reveals that the highest mutation load is associated with chronic sun exposure, and that the PREX2 gene is mutated in approximately 14 per cent of cases
- Michael F. Berger
- , Eran Hodis
- & Levi A. Garraway
 MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis
MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis