
Abstract
Extract: We are reporting a patient with classic, thiamine-unresponsive maple syrup urine disease (MSUD) associated with severe fasting hypoglycemia. The potential gluconeogenic amino acids, with the exception of glutamate, were markedly depressed. Glutamine and alanine concentrations increased rapidly with the concomitant fall in branched chain amino acids following initiation of a diet free of leucine, isoleucine, and valine. Intravenous glucagon and oral fructose resulted in normal glycemic responses (Δ76 and Δ40 mg/100 ml, respectively) and no significant rise in blood lactate or pyruvate.
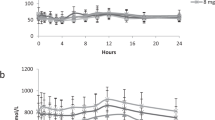
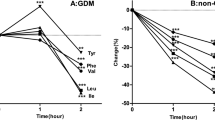
During two fasting periods (day 17 and 49) when the branched chain amino acids were elevated, hypoglycemia ensued within 9 hr (47 and 44 mg/100 ml, respectively). Blood lactate and alanine decreased, and blood ketone bodies rose rapidly with fasting. Plasma insulin was undetectable (< 5 μUU/ml) in the face of hypoglycemia, plasma growth hormone was detectable at all times, and plasma cortisone increased with fasting. At the time of the third fast (day 58), when branched chain amino acids were in the normal range, blood glucose was maintained for 13 hr (62 mg/100 ml). Ketone bodies increased but lactate concentration did not change. Plasma alanine concentration was within the normal range (354 μUm) and fell during the fast.
Both before and after specific dietary therapy, alanine infusions resulted in no rise in plasma glucose. Plasma insulin remained < 5 μUU/ml during each infusion. With each infusion, regardless of the plasma branched chain amino acid concentrations, there was a transient rise in glutamate and a marked and sustained increase in glutamine. Hyperlactic and hyperpyruvic acidemia were not observed. Concentrations of valine, isoleucine, α-ketoisocaproic acid, α-ketoisovaleric acid, or α-keto-β-methyl valeric acid 10-fold greater than that present in the patient's plasma or in those reported in other patients with MSUD had little or no effect on the in vitro activity of bovine liver glutamate dehydrogenase, an enzyme which serves as a major link between carbohydrate and amino acid metabolism. In contrast, there was a 22–42% stimulation of glutamate dehydrogenase activity with concentrations of leucine similar to that found in our patient's plasma.
Speculation: Hypoglycemia associated with MSUD appears to be related to a defect in gluconeogenesis from amino acids, which cannot be accounted for by abnormalities of the ratelimiting gluconeogenic enzymes or hyperinsulinemia. It would appear that there is a preferential shunting of 3-carbon substrates from amino acids into glutamine leading to decreased net oxaloacetate production and impaired gluconeogenesis.
Similar content being viewed by others

Article PDF
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Haymond, M., Karl, I., Feigin, R. et al. Hypoglycemia and Maple Syrup Urine Disease: Defective Gluconeogenesis. Pediatr Res 7, 500–508 (1973). https://doi.org/10.1203/00006450-197305000-00003
Issue Date:
DOI: https://doi.org/10.1203/00006450-197305000-00003
Keywords
This article is cited by
-
Interrupting the mechanisms of brain injury in a model of maple syrup urine disease encephalopathy
Journal of Inherited Metabolic Disease (2012)
-
Maple syrup urine disease: An uncommon cause for neonatal metabolic distress
Indian Journal of Clinical Biochemistry (1999)
-
Hypoglycaemia in classical maple syrup urine disease is not due to hyperinsulinism
Journal of Inherited Metabolic Disease (1983)
-
Mild variant of maple syrup urine disease
European Journal of Pediatrics (1976)
