
Abstract
Nitric oxide and excitatory amino acids contribute to hypoxic-ischemic brain injury. Agmatine, an endogenous neurotransmitter or neuromodulator, is an inhibitor of nitric oxide synthase and an antagonist of N-methyl-d-aspartate receptors. Does agmatine reduce brain injury in the rat pup hypoxic-ischemic model? Seven-day old rat pups had right carotid arteries ligated followed by 2.5 h of hypoxia (8% oxygen). Agmatine or vehicle was administered by i.p. injection at 5 min after reoxygenation and once daily thereafter for 3 d. Brain damage was evaluated by weight deficit of the right hemisphere at 22 d after hypoxia by a blinded observer. Agmatine treatments significantly reduced weight loss in the right hemisphere from −30.5 ± 3.6% in vehicle-treated pups (n = 22) to −15.6 ± 4.4% in the group treated with 50 mg/kg (n = 18, p < 0.05) and to −15.0 ± 3.7% in the group treated with 100 mg/kg (n = 18, p < 0.05), but the group treated with 150 mg/kg showed no reduction. Other pups received agmatine or vehicle at 5 min after reoxygenation, and brain biochemistry was assessed. Levels of endogenous brain agmatine rose 2- to 3-fold owing to hypoxic-ischemic (3 h), whereas pups treated with agmatine (100 mg/kg) showed 50-fold higher brain agmatine levels (3 h). Agmatine (100 mg/kg) blocked a hypoxia-induced increase in brain nitric oxide metabolites at 6 h (vehicle-treated, +60.2 ± 15.2%; agmatine-treated, +4.2 ± 8.4%;p < 0.05). Agmatine thus reduces brain injury in the neonatal rat hypoxic-ischemic model, probably by blunting the rise in nitric oxide metabolites normally seen after hypoxia.
Similar content being viewed by others

Main
Nitric oxide (NO) and excitatory amino acids contribute to ischemic brain injury. Inhibitors of nitric oxide synthase (NOS) and antagonists of N-methyl-d-aspartate (NMDA) glutamate receptors are neuroprotective in hypoxia-ischemic (HI) brain injury (1–10). Formed from the decarboxylation of l-arginine by the enzyme arginine decarboxylase (11–14), agmatine is a 4-carbon elongated relative of the synthetic neuroprotective agent, aminoguanidine (12). Agmatine has long been known to exist in plants, bacteria, insects, and invertebrates (13). Only recently was it realized that agmatine is synthesized in mammalian brain (11), where agmatine acts as both a neurotransmitter and neuromodulator (12, 14–18). From distribution studies of arginine decarboxylase in primary cell cultures, agmatine is thought to be synthesized predominantly by astroglia cells, then released and taken up into neurons by active transport (12, 14). Agmatine possesses modest affinities for various receptors, including as an inhibitor of the NMDA subclass of glutamate receptors (19–21). Agmatine also inhibits all isoforms of NOS (22–25), with highest reported activity (Ki = 29 μM) as an irreversible inactivator of neuronal NOS (25). Treatments with agmatine are neuroprotective in vitro(21, 22), as well as in gerbil ischemic brain injury, adult rat ischemic brain injury (26), and adult rat spinal cord injury (16, 27, 28). The effects of agmatine in neonatal HI brain injury have not previously been tested, nor has correlation been studied between the neuroprotective properties of agmatine and NO production in the brain.
HI brain injury is a serious cause of death and disability in human newborns. The developmental stage of the brain of the 7-d-old rat pup resembles that of newborn humans (29). As described for the Rice-Vannucci-Brierley rat pup HI model (30), HI injury may best match the injury caused by birth asphyxia in full-term human infants. The neonatal rat HI model (30) has been well characterized and extensively used to assess synthetic neuroprotective agents (1–3, 8–10, 29, 31, 32), including aminoguanidine (2, 3). We have used the HI model to evaluate the neuroprotective potency of several new drugs (33, 34). The purpose of the present study was thus to evaluate the effects of agmatine on brain injury and NO production by using the neonatal rat HI model.
METHODS
Animal protocol.
This protocol was approved by our institutional committee on animal use. Rats were cared for in accordance with National Institutes of Health guidelines. The neonatal rat HI procedure was performed as described by Rice et al.(30). Seven-day-old Sprague-Dawley (Harlan Sprague-Dawley, Indianapolis, IN, U.S.A.) rat pups of either sex, weighing between 12 and 17 g, were anesthetized with isoflurane (4% induction, 2% maintenance). The right common carotid artery was exposed, isolated, and permanently doubly ligated. After surgery, the rat pups were returned to their dams for 2–3 h recovery. Complete hypoxic exposure was achieved by placing the rat pups in 1.5-L sealed jars surrounded by a 37°C water bath and subjected to a humidified mixture of 8% oxygen–92% nitrogen delivered at 4 L/min for 2.5 h. After this hypoxic exposure, some pups were returned to their dams and allowed to recover and grow for 22 d. Pups were weighed before injury and again at 4, 7, 11, 14, and 22 d after injury.
Drug treatment.
Pups from each litter were randomly assigned, and marked, to a control group or for treatment with agmatine sulfate salt (Sigma Chemical Co., St. Louis, MO, U.S.A.). Agmatine in doses of 50, 100, or 150 mg/kg was dissolved in 0.9% saline and administered by i.p. injection at 5 min after hypoxia exposure and once daily thereafter for 3 d. The control group was given 0.9% saline alone. These doses were chosen as the most promising from our pilot experiments.
Measurement of rectal temperature.
To evaluate whether neuroprotection by agmatine was dependent on systemic hypothermia, rectal temperature was measured with a 36-gauge flexible thermocouple (Omega Engineering Inc., Stamford, CT, U.S.A.). This was performed in a subset of pups (six from the control group and six given 100 mg/kg of agmatine at 5 min after hypoxia exposure) before placing the pups in the jar and at 0.25, 0.5, 1, 2, 4, 6, and 24 h after removal.
Gross neuropathologic grading.
Rat pups were anesthetized with pentobarbital and decapitated 22 d after hypoxic exposure. The brains were removed, scored, and weighed by a observer blind to the code. Brains were scored normal, mild, moderate, or severe by the method of Palmer et al.(29). “Normal” meant no reduction in the size of the right hemisphere, “mild” meant visible reduction in right hemisphere size, “moderate” meant large reduction in hemisphere size with a visible infarct in the right parietal area, and “severe” meant near total destruction of the hemisphere (29). After removing the cerebellum and brainstem, the brain was divided into two hemispheres and weighed. Results are presented as the percent loss of hemispheric weight of the right side relative to the left [(left − right)/left × 100]. This HI model results in brain damage only on the ipsilateral side (29, 30). The loss of hemispheric weight can be used as a measure of brain damage in this model, inasmuch as enough time elapsed to allow resorption of the dead tissue (8, 31). Also the brain of the newborn rat is growing rapidly, and the dead tissue does not contribute to this growth.
Measurement of NO metabolites.
A second set of experiments was performed to determine the effect of agmatine on nitrite and nitrate levels. The same neonatal HI procedure was performed using the 2.5-h period of hypoxia. Pups were randomly assigned to a sham group (n = 5), treatment with saline (n = 26), or treatment with agmatine at a dose of 100 mg/kg (n = 25) at 5 min after hypoxia, and sampled at 1, 3, 4.5, 6, 12, and 24 h after hypoxia. Because NO is a labile molecule, we used a reported method (35) to measure the stable end products of NO metabolism: nitrites and nitrates. At each time, the pups were decapitated, brains were removed, and each side of the cerebral cortex was frozen at −80°C. Brain samples were later homogenized in 4 vol of ice-cold buffer (0.1 M potassium phosphate, pH 7.5, 20 mM EDTA) and centrifuged at 20,000 ×g for 30 min at 4°C. Total nitrate and nitrite were determined using nitrate reductase from Escherichia coli to convert nitrate to nitrite (verified for each batch, using a sodium nitrate standard, that all nitrate was converted to nitrite). The concentration of nitrite was measured colorimetrically using the Griess reaction (35). Sodium nitrite was used as the standard. The data are expressed as nanomoles of nitrite/nitrate per milligram protein. Protein was assayed using the method of Bradford (36). Nitrite/nitrate in the sham rat pup brain was used as the baseline level. Nitrate/nitrite in the right (ipsilateral) cortex of agmatine-treated and control groups are presented as the percent increase in the right hemisphere relative to the left hemisphere [(right − left)/left × 100].
Measurement of agmatine levels.
Agmatine levels in brains of naïve pups and HI rat pups treated with saline or agmatine were measured according to our published HPLC method (37) with minor modifications. Neonatal ischemia (ligation), hypoxia (8% O2), and drug treatments were as described above, and the brains were immediately processed at 1 h during hypoxia, or 5 min, 1, 3, 4.5, 6, and 24 h after completion of 2.5 h of hypoxia. Brain samples were homogenized in 4 vol of ice-cold buffer (0.1 M potassium phosphate, pH 7.5, 20 mM EDTA) and centrifuged at 20,000 ×g for 30 min at 4°C. An internal standard, 1-(3-aminopropyl)2-pipecodine (Aldrich Chemical Company, Milwaukee, WI, U.S.A.) was added to each sample (0.8 ng/per injection). The supernatants were mixed with equal volumes of ice-cold 10% trichloroacetic acid and centrifuged again. Aliquots (20 μL) were mixed with 6 μL of 1 M NaOH and 20 μL of the O-phthalaldehyde/mercapto-ethanol derivatizing reagent for 2 min, and then immediately injected (20 μL) into the HPLC system. The HPLC system consisted of a pump and multisolvent delivery system (Waters model 600; Marlborough, MA. U.S.A.), a Gilson model 121 fluorescence detector (excitation wavelength of 350 nm and emission wavelength of 450 nm), and a 5-μm Hypersil ODS, 150 × 4.6 mm I.D. column (Supelco Inc. Bellefonte, PA, U.S.A.). The isocratic mobile phase consisted of a mixture of 75 mL of 0.1 M potassium phosphate (pH 5.7), 50 mL of 75 mM octyl sulfate sodium salt, 300 mL of methanol, 200 mL of acetonitrile, and 375 mL of water with a flow rate of 1.0 mL/min. Peak heights were used to quantify results from a standard curve.
Statistics.
Categorical variables were analyzed with the χ2 test. Continuous variables (brain weight, NO metabolites, and agmatine brain levels) are presented as mean ± SEM, and the statistical significance of differences among groups were determined using ANOVA with the Student-Newman-Keuls test. Repeated measures ANOVA was used for rectal temperature and body weight. Differences were considered significant at p < 0.05.
RESULTS
Rectal temperatures obtained before placing the pups in the 8% O2 and at 0.25, 0.5, 1, 2, 4, 6, and 24 h after hypoxia were not significantly different among any groups at any times (Fig. 1). This finding tends to rule out an indirect hypothalamic effect of agmatine (15). Also, no pups died during the 22-d recovery period after hypoxia in agmatine-treated or saline-treated groups. Body weights of the treated groups were not significantly different from control pups before injury or at 4, 7, 11, 14, or 22 d after injury. Body weights increased significantly with time in all groups as the pups grew.

Effect of agmatine on rectal temperatures in the HI rat pups. Administration of agmatine (100 mg/kg) or vehicle i.p. occurred at 5 min after hypoxia. There were no significant differences between groups during the various times.
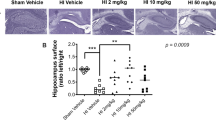
The proportion of pup brains scored as damaged (moderate and severe) is shown in Figure 2. Agmatine decreased the percentage of brain scored as damaged from 59.1% in the vehicle group to 22.2% with 100 mg/kg (p < 0.05). The dose of 50 mg/kg of agmatine produced a similar effect (27.8%), but this was not statistically significant. The dose of 150 mg/kg had no effect (58.8%, p > 0.05).

The effect of different doses (50–150 mg/kg i.p.) of agmatine administered at 5 min after HI on the degree of brain damage after 22 d. Brain damage was scored by a blinded observer 22 d after HI. Posttreatment with 100 mg/kg of agmatine decreased the number of brains scored as damaged compared with vehicle (*p < 0.05).
Left hemisphere weights were 499.4 ± 6.7 mg (n = 22), 500.1 ± 9.1 mg (n = 18), 519.6 ± 8.7 mg (n = 18), and 509.0 ± 7.7 mg (n = 17) in the 0 (vehicle), 50, 100, and 150 mg/kg agmatine-treatment groups, respectively. Thus, the left hemisphere was unaffected (p > 0.05) by the HI procedure, as expected (29). The percent reduction in right hemispheric weight is shown in Figure 3. Agmatine significantly reduced the decrease in right hemisphere weight from −30.5 ± 3.6% in the vehicle group (n = 22) to −15.6 ± 4.4% in the group receiving 50 mg/kg of agmatine (n = 18, p < 0.05) and to −15.0 ± 3.7% in the group receiving 100 mg/kg of agmatine (n = 18, p < 0.05). Treatment with 150 mg/kg of agmatine did not effect the reduction in right hemispheric weight (−33.0 ± 5.7%, n = 17) compared with the vehicle (p > 0.05).

The dose-response for percentage reduction in right cerebral hemisphere weight measured using the left hemisphere weight as the standard. Treatment was given 5 min after hypoxia, and injury was evaluated 22 d later. Posttreatment with 50 and 100 mg/kg of agmatine decreased the percentage reduction in right hemisphere weight compared with vehicle (*p < 0.05), with a peak effect at 100 mg/kg.
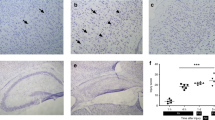
Total NO metabolites are shown in Figure 4. HI resulted in a significant increase in NO metabolites in the right cortex at 6 h after hypoxia compared with the sham group (p < 0.05). Agmatine (100 mg/kg) eliminated the increase in NO metabolites at 6 h after hypoxic exposure (vehicle, +60.2 ± 15.2%, n = 5; agmatine, +4.2 ± 8.4%, n = 6;p < 0.05). There were no other statistically significant differences in NO metabolites at 1, 3, 4.5, 12, and 24 h after hypoxia.

Total nitrate/nitrite production in the contralateral and the ipsilateral hemisphere cortex of agmatine-treated and control groups was measured at 1, 3, 4.5, 6, 12, and 24 h after hypoxia. The pups were treated with 100 mg/kg of agmatine or vehicle at 5 min after hypoxia. Data are presented as the percent increase of the right hemisphere relative to the left hemisphere [(right − left)/left × 100]. Hypoxia results in an increase in NO metabolites in the right cortex at 6 h after hypoxia that is eliminated by treatment with agmatine. *p < 0.05, significant vs agmatine-treated group at the same time; #p < 0.05, significant vs the sham group.
Agmatine levels in pup brain cortex are shown in Table 1. During hypoxic exposure for 1 h (without injected agmatine), a tendency was noted for a rise in endogenous agmatine concentration in the ipsilateral cortex (Table 1). Likewise, posthypoxic (2.5 h) pups treated with only saline for 5 min, 1 h, and 3 h showed significant elevations in endogenous agmatine levels (Table 1;p < 0.01 versus naïve pups). Thus, hypoxia alone caused a modest 2- to 3-fold rise (p < 0.01) in cortical agmatine. On top of this, as expected, injection of 100 mg/kg of agmatine i.p. markedly increased brain agmatine levels (p < 0.01). Cortical agmatine levels were 56-fold higher after 3 h, and declined to 4-fold higher by 24 h, than levels in naïve pups, after the 100 mg/kg treatment (Table 1).
DISCUSSION
This study demonstrates that administration of agmatine at 5 min after hypoxia and then once a day thereafter for 3 d significantly reduces the degree of brain injury in the focal ischemia model of the neonatal rat pup. Similar results with agmatine have been reported in the adult rat focal cerebral ischemia model (26), the adult gerbil global forebrain ischemia model (26), and the adult rat spinal cord injury model (16, 27, 28). Delayed neuronal injury sometimes requires a prolonged period to develop (32). Therefore, we extended the time from injury to brain assessment from the 72 h used in the study by Gilad et al.(26) of adult rats to 22 d. There was no reduction in the efficacy of agmatine over this period. Thus, agmatine is effective at doses of 50 and 100 mg/kg in the rat pup, which is similar to the doses shown to be effective in adult rat focal cerebral ischemia and in adult gerbil global forebrain ischemia (26) and in adult rat spinal cord injury (16, 27, 28). In our hands, the maximally effective dose of agmatine appeared at 100 mg/kg in rat pups. However, unlike in the adult rat spinal cord injury model (16, 27), a dose of 150 mg/kg of agmatine showed no neuroprotective effect in our study. The reason for this difference is unclear but may relate to differences in animal age or other variation in the protocols. Other neuroprotective agents have been described with U-shaped dose-response curves (38, 39). Further investigation of the loss of efficacy of agmatine at high doses is warranted.
Posttreatment with 100 mg/kg of agmatine had no effect on body temperature in the rat pups (Fig. 1). The neuroprotection offered by agmatine therefore does not depend on systemic hypothermia. Furthermore, agmatine treatment seems to be of low toxicity. Even with the high agmatine dose (150 mg/kg), growth was not adversely affected.
NO is enzymatically formed from the terminal guanidino-nitrogen of l-arginine by NOS. NOS has three isoforms. NOS I, formerly neuronal NOS, is constitutively expressed largely in neurons in the brain and activated by intracellular Ca2+. NOS II, formerly inducible NOS, is not present in normal tissue but is induced at the transcriptional level in glia and inflammatory cells by inflammation and tissue injury. NOS III, formerly endothelial NOS, is constitutively expressed in endothelial cells in the brain and is important in vasodilation (40). HI increases NO production acutely by NOS I and III, and after a delay by NOS II (40). Inhibition of NOS III worsens injury to the brain in stroke models by reducing brain blood flow (41). Inhibition of NOS I in adult stroke models by gene knockout in adult mice (41) or by specific inhibitor in adult rats (42) reduces injury. Furthermore, inhibiting NOS I in newborn HI brain injury by gene knockout (43) in newborn mice reduces injury. Similarly inhibition of NOS II in adult stroke models by gene knockout in mice (44) or specific inhibitor in rats (4) reduces injury. Inhibiting NOS II by specific inhibitor in the newborn rat model of HI also reduces injury (2). Even nonselective NOS inhibitors have shown efficacy in the rat pup (1, 5). Agmatine is more than an order of magnitude more potent with NOS I and NOS II than with NOS III (23). Also, agmatine significantly suppresses lipopolysaccharide-induced NO production in a concentration-dependent manner in vitro(14, 24), and agmatine also reduces neuronal death caused by microglia-derived NO (24). Agmatine has been reported to irreversibly inactivate neuronal NOS (25).
In the present study, HI rat pups showed elevations in NO metabolite concentrations in the brain during reperfusion 6 h after hypoxia, and agmatine treatment (100 mg/kg) eliminated this increase in NO metabolites. The results indicate that agmatine suppresses NO production after hypoxia in vivo as is known to occur in vitro(22–25). Reduction of NO generation may therefore be one mechanism of neuroprotection by agmatine. However, the effect of HI on total nitrates and nitrites is complex. Higuchi et al.(3) have reported a biphasic increase in NO metabolites in the neonatal rat HI model. The first increase in NO production occurred 1 h into the hypoxic period, caused by neuronal NOS, and resolved by the end of the hypoxic period (3). A secondary peak occurred at 3 h after hypoxia, when their last measurement was taken, which was blocked with inducible NOS inhibitors or by blocking the primary peak. Likewise, our previous studies have shown these two phases of increase in NO metabolites, with the secondary peak seen at 6 h after hypoxia (33).
NO production may induce DNA fragmentation by damage to mitochondria, resulting in loss of transmembrane potential, induction of a mitochondrial transition pore, release of cytochrome c from mitochondria, and initiation of caspase activation, thereby initiating the apoptotic cascade (45). NO, through the production of peroxynitrite, can also cause oxidative DNA damage and single-stranded DNA breaks and initiate cellular injury through stimulation of poly(ADP-ribose) polymerase (46) and ATP depletion. Loss of calcium homeostasis in neuronal cells is also considered a key event in HI-induced brain damage. When oxygenation fails, ATP formation drops and excessive calcium influx occurs through voltage-sensitive calcium channels, leading to the production of excitatory amino acids such as glutamate. This causes activation of NMDA receptors, leading to further calcium influx, and triggering numerous and very complex cellular changes leading to cell damage. As neuroprotective agents, NMDA receptor antagonists also clearly remain potential therapeutic drugs (5–10). Agmatine has been reported to selectively block the NMDA subclass of glutamate receptors (19–21). Agmatine also is neuroprotective in cultured cerebellar granule cells and PC12 cells (20, 21). Therefore, besides blocking NO production, a second mechanism responsible for the neuroprotective effects of agmatine may involve selective blockade of the NMDA receptor.
Endogenous cortical agmatine in naïve pups was found to be 0.138 ± 0.02 μg/g wet weight. This concentration is slightly lower than that previously reported in naïve adult rat brain by us (0.331–1.105 μg/g tissue) (37) and by others (0.2–0.4 μg/g tissue) (11), and in rat spinal cord (0.56–0.96 μg/g tissue) (16). HI alone increased (p < 0.01) cortical levels of endogenous agmatine 2- to 3-fold (Table 1). This finding indicates that agmatine levels may be endogenously regulated. Three hours after administration of agmatine at the dose of 100 mg/kg, a massive increase in brain agmatine to 7.69 ± 0.58 μg/g wet cortex occurred. This suggests that agmatine is readily transported into the brain in this model. Thus, the effective concentration of agmatine in suppressing NO production and in reducing brain damage was 4–7 μg/g wet weight in the neonatal rat HI model. This concentration stands some 40 times higher than endogenous agmatine concentrations in the sham pup brains. On the assumption that 1 g of wet weight of tissue corresponds to 1 mL of water, the endogenous concentration of agmatine in brain can be estimated at 59.2 μM. In this case, 100 mg/kg i.p. agmatine treatment yields levels of agmatine within the range reported to suppress microglial NO production (24).
CONCLUSIONS
In conclusion, our findings indicate that the endogenous amine, agmatine, has neuroprotective properties. Treatment with agmatine reduces brain injury in the neonatal rat HI model. Suppression of NO production after hypoxia by agmatine is a potential mechanism of this neuroprotection. Our results substantiate earlier data that agmatine is a novel and potentially advantageous therapeutic agent for the treatment of brain injury (16, 26–28).
Abbreviations
- NO:
-
nitric oxide
- NOS:
-
nitric oxide synthase
- HI:
-
hypoxia-ischemia, hypoxic-ischemic
- NMDA:
-
N-methyl-d-aspartate
References
Hamada Y, Hayakawa T, Hattori H, Mikawa H 1994 Inhibitor of nitric oxide synthesis reduces hypoxic-ischemic brain damage in the neonatal rat. Pediatr Res 35: 10–14
Tsuji M, Higuchi Y, Shiraishi K, Kume T, Akaike A, Hattori H 2000 Protective effect of aminoguanidine on hypoxic-ischemic brain damage temporal profile of brain nitric oxide in neonatal rat. Pediatr Res 47: 79–83
Higuchi Y, Hattori H, Kume T, Tsuji M, Adaike A, Furusho K 1998 Increase in nitric oxide in the hypoxic-ischemic neonatal rat brain suppression by 7-nitroindazole aminoguanidine. Eur J Pharmacol 342: 47–49
Iadecola C, Zhang F, Xu X 1995 Inhibition of inducible nitric oxide synthase ameliorates cerebral ischemic damage. Am J Physiol 268: R286–R292
Ashwal S, Cole DJ, Osborne S, Osborne TN, Pearce WJ 1995 L-NAME reduces infarct volume in a filament model of transient middle cerebral artery occlusion in the rat pup. Pediatr Res 38: 652–656
Hagberg H 1992 Hypoxic-ischemic damage in the neonatal brain: excitatory amino acids. Dev Pharmacol Ther 18: 139–144
Olney JW, Ikonomidou C, Mosinger JL, Friedrich G 1989 MK-801 prevents hypoxic-ischemic neuronal degeneration in infant rat brain. J Neurosci 9: 1701–1704
Hagberg H, Gilland E, Diemer NH, Andiné P 1994 Hypoxia-ischemia in the neonatal rat brain: histopathology after post-treatment with NMDA non-NMDA receptor antagonists. Biol Neonate 66: 205–213
Gilland E, Puka-Sundvall M, Andiné P, Bona E, Hagberg H 1994 Hypoxic-ischemic injury in the neonatal rat brain: effects of pre- post-treatment with the glutamate release inhibitor BW1003C87. Dev Brain Res 83: 79–84
Cai Z, Xiao F, Fratkin JD, Rhodes PG 1999 Protection of neonatal rat brain from hypoxic-ischemic injury by LY379268, a group II metabotropic glutamate receptor agonist. Neuroreport 10: 3927–3931
Li G, Regunathan S, Barrow CJ, Eshraghi J, Cooper R, Reis DJ 1994 Agmatine: an endogenous clonidine-displacing substance in the brain. Science 263: 966–969
Regunathan S, Reis DJ 2000 208 Characterization of arginine decarboxylase in rat brain liver: distinction from ornithine decarboxylase. J Neurochem 74: 2201–2208
Tabor CW, Tabor H 1984 Polyamines. Annu Rev Biochem 53: 749–790
Reis DJ, Regunathan S 2000 Is agmatine a novel neurotransmitter in brain?. Trends Pharmacol Sci 21: 187–193
Kalra SP, Pearson E, Sahu A, Kalra PS 1995 Agmatine, a novel hypothalamic amine, stimulates pituitary luteinizing hormone release in vivo hypothalamic luteinizing hormone-releasing hormone release in vitro. Neurosci Lett 194: 165–168
Fairbanks CA, Schreiber KL, Brewer KL, Yu CG, Stone LS, Kitto KF, Nguyen HO, Grocholski BM, Shoeman DW, Kehl LJ, Regunathan S, Reis DJ, Yezierski RP, Wilcox GL 2000 Agmatine reverses pain induced by inflammation, neuropathy, spinal cord injury. Proc Natl Acad Sci USA 97: 10584–10589
Blantz RC, Satriano J, Gabbai F, Kelly C 2001 Biological effects of arginine metabolites. Acta Physiol Scand 168: 21–25
Raasch W, Schafer U, Chun J, Dominiak P 2001 Biological significance of agmatine, an endogenous ligand at imidazoline binding sites. Br J Pharmacol 133: 755–780
Yang XC, Reis DJ 1999 Agmatine selectively blocks the N-methyl-d-aspartate subclass of glutamate receptor channels in rat hippocampal neurons. J Pharmacol Exp Ther 288: 544–549
Olmos G, DeGregorio-Rocasolano N, Regalado MP, Gasull T, Boronat MA, Trullas R, Villarroel A, Lerma J, García-Sevill JA 1999 Protection by imidazol(ine) drugs agmatine of glutamate-induced neurotoxicity in cultured cerebellar granule cell through blockade of NMDA receptor. Br J Pharmacol 127: 1317–1326
Regunathan S, Zhou P, Reis DJ 2000 Agmatine protects against cell death induced by NMDA serum deprivation but not by protein kinase inhibition or calcium entry. Soc Neurosci Abstr 26: 2171( abstr)
August M, Viossat I, Marin JG, Chabrier PE 1995 Selective inhibition of inducible nitric oxide synthase by agmatine. Jpn J Pharmacol 69: 285–287
Galea E, Regunathan S, Eliopoulos V, Feinstein DL, Reis DJ 1996 Inhibition of mammalian nitric oxide synthase by agmatine, an endogenous polyamine formed by decarboxylation of arginine. Biochem J 316: 247–249
Abe K, Abe Y, Saito H 2000 Agmatine suppresses nitric oxide production in microglia. Brain Res 872: 141–148
Demady DR, Jianmongkol S, Vuletich JL, Bender AT, Osawa Y 2001 Agmatine enhances the NADPH oxidase activity of neuronal NO synthase leads to oxidative inactivation of the enzyme. Mol Pharmacol 59: 24–29
Gilad GM, Salame K, Rabey JM, Gilad VH 1996 Agmatine treatment is neuroprotective in rodent brain injury models. Life Sci 58: PL41–PL46
Yu CG, Marcillo AE, Fairbanks CA, Wilcox GL, Yezierski RP 2000 207 Agmatine improves locomotor function reduces tissue damage following spinal cord injury. Neuroreport 11: 3203–3207
Gilad GM, Gilad VH 2000 Accelerated functional recovery neuroprotection by agmatine after spinal cord ischemia in rats. Neurosci Lett 296: 97–100
Palmer C, Vannucci RC, Towfighi J 1990 Reduction of perinatal hypoxic-ischemic brain damage with allopurinol. Pediatr Res 27: 332–336
Rice JE, Vannucci RC, Brierley JB 1981 The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9: 131–141
Bona E, Johansson BB, Hagberg H 1997 Sensorimotor function neuropathology five six weeks after hypoxia-ischemia in seven-day-old rats. Pediatr Res 42: 678–683
Trescher WH, Ishiwa S, Johnston MV 1997 Brief post-hypoxic-ischemic hypothermia markedly delays neonatal brain injury. Brain Dev 19: 326–338
LeBlanc MH, Feng Y, Fratkin JD 2000 N-tosyl-l-phenylalanyl-chloromethylketone reduces hypoxic-ischemic brain injury in rat pups. Eur J Pharmacol 390: 249–256
Feng YZ, LeBlanc MH 2000 GPI-6150, a potent PARP inhibitor, does not affect the severity of hypoxic-ischemic brain injury in newborn rat. Soc Neurosci Abstr 26: 773( abstr)
Granger JP, Kassab S, Novak J, Reckelhoff JF, Tucker B, Miller T 1999 Role of nitric oxide in modulating renal function arterial pressure during chronic aldosterone excess. Am J Physiol 276: R197–R202
Bradford MM 1976 A rapid sensitive method for quantitation of microgram quantities of protein utilizing the principle of protein dye binding. Anal Biochem 72: 248–254
Feng Y, Halaris AE, Piletz JE 1997 Determination of agmatine in brain plasma using high- performance liquid chromatography with fluorescence detection. J Chromatogr B 691: 277–286
Omar BA, McCord JM 1990 The cardioprotective effects of Mn-superoxide dismutase is lost at high doses in the postischemic isolated rabbit heart. Free Radic Biol Med 9: 473–478
Takhashi K, Pieper AA, Croul SE, Zhang J, Snyder SH, Greenberg JH 1999 Post-treatment with an inhibitor of poly(ADP-ribose) polymerase attenuates cerebral damage in focal ischemia. Brain Res 829: 46–54
Samdani AF, Dawson TM, Dawson VL 1997 Nitric oxide synthase in models of focal ischemia. Stroke 28: 1283–1288
Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA 1994 Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science 265: 1883–1885
Escott JK, Beech JS, Haga KK, Williams SCR, Meldrum BS, Bath PMW 1998 Cerebroprotective effect of the nitric oxide synthase inhibitors, 1-(2-trifluoromethylphenyl) imidazole 7-nitro indazole, after transient focal cerebral ischemia in the rat. J Cereb Blood Flow Metab 18: 281–287
Ferriero DM, Holtzman DM, Black SM, Sheldon RA 1996 Neonatal mice lacking neuronal nitric oxide synthase are less vulnerable to hypoxic-ischemic injury. Neurobiol Dis 3: 64–71
Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME 1997 Delayed reduction of ischemic brain injury neurological deficits in mice lacking the inducible nitric oxide synthase gene. J Neurosci 17: 9157–9164
Budd SL 1998 Mechanisms of neuronal damage in brain hypoxia/ischemia: focus on the role of mitochondrial calcium accumulation. Pharmacol Ther 80: 203–229
Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA 1997 Ischemic brain injury is mediated by the activation of poly(ADP-Ribose)polymerase. J Cereb Blood Flow Metab 17: 1143–1151
Author information
Authors and Affiliations
Corresponding author
Additional information
Supported in part by an unrestricted grant from Burroughs Wellcome Company and in part by National Institute of Mental Health grant 57601.
Rights and permissions
About this article
Cite this article
Feng, Y., Piletz, J. & Leblanc, M. Agmatine Suppresses Nitric Oxide Production and Attenuates Hypoxic-Ischemic Brain Injury in Neonatal Rats. Pediatr Res 52, 606–611 (2002). https://doi.org/10.1203/00006450-200210000-00023
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-200210000-00023
This article is cited by
-
Maintenance of the Neuroprotective Function of the Amino Group Blocked Fluorescence-Agmatine
Neurochemical Research (2021)
-
Therapeutic Effect of Agmatine on Neurological Disease: Focus on Ion Channels and Receptors
Neurochemical Research (2019)
-
Decreased plasma agmatine levels in autistic subjects
Journal of Neural Transmission (2018)
-
Agmatine Induces Nrf2 and Protects Against Corticosterone Effects in Hippocampal Neuronal Cell Line
Molecular Neurobiology (2015)
-
Protective effects of agmatine against d-galactosamine and lipopolysaccharide-induced fulminant hepatic failure in mice
Inflammopharmacology (2014)
