
Abstract
The role of superoxide radical formation in the pathogenesis of perinatal hypoxic-ischemic injury was examined using transgenic (Tg) mice expressing three times normal amounts of copper/zinc-superoxide dismutase (CuZn/SOD). Fourteen litters of postnatal d 7 strain 218/3 mice were subjected to right common carotid artery ligation followed by 90 min of hypoxia in an 8% oxygen/humidified chamber maintained at 37°C. Both Tg mice (n = 32) and their nontransgenic (nTg) littermates (n = 30) survived the injury equally. Evaluation of infarcted brain areas measured by video image analysis of three coronal brain sections through the anterior hippocampus from each animal revealed that the Tg animals suffered brain infarction more frequently than did nTg mice. Blinded histologic scoring of cerebral cortex and striatum 5 d after injury revealed that Tg mice were more likely to have higher histologic severity scores than their nTg littermates (p = 0.0463, Mann-Whitney U test). These findings suggest that brain injury in perinatal hypoxia-ischemia may be mediated in part by free radical formation from excessive hydrogen peroxide or nitric oxide production.
Similar content being viewed by others
Main
Perinatal cerebral hypoxia-ischemia, resulting from compromised placental or pulmonary gas exchange, is a major cause of acute perinatal brain injury, leading ultimately to neurologic damage such as cerebral palsy, mental retardation, and epilepsy(1). To maintain adequate brain metabolism during hypoxia, vasodilation occurs. If asphyxia persists, however, blood flow to the brain is ultimately reduced due to secondary hypotension and bradycardia, resulting in cerebral ischemia and necrosis(2).
During cerebral hypoxia-ischemia, a number of metabolic changes occur that result in the production of oxygen free radicals, most notably superoxide. The accumulation of oxygen free radicals can cause neuronal death through reactions involving lipid peroxidation(3, 4), protein oxidation(5, 6), and DNA damage(7, 8).
CuZn/SOD is an endogenous antioxidant enzyme that removes free radicals from neurons in the brain by converting superoxide anion to hydrogen peroxide. Glutathione peroxidase and catalase are then able to catalyze the disposal of hydrogen peroxide to H2O and O2. Although CuZn/SOD has been shown to reduce infarction severity induced by certain types of injury in adult animal models(9–15), its neuroprotective role in the developing perinatal brain to hypoxic-ischemic injury has yet to be studied.
In perinatal animal models, in vivo studies have demonstrated that the hydroxyl radical scavengers allopurinol and oxypurinol reduce infarction severity due to hypoxic-ischemic injury implying that oxygen free radicals are partly responsible for the damage caused during perinatal cerebral hypoxia-ischemia(16, 17). Because CuZn/SOD is a catalyst of free radical turnover, it might also lessen brain damage during perinatal hypoxia-ischemia, as do the oxygen free radical scavengers allopurinol and oxypurinol.
We used a strain of Tg mice whose heterozygotes express a 3.1-fold increase in human SOD-1 activity in all brain regions compared with their nTg litter mates(18). By using a model of hypoxic-ischemic encephalopathy and comparing hypoxic-ischemic brain damage in CuZn/SOD Tg mice and nTg mice, we sought to determine the effect of overexpression of the CuZn/SOD gene on perinatal mice undergoing hypoxic-ischemic insult.
METHODS
Tg mice. The mice used for these experiments were bred as follows. Tg mice of strain TgHS/SF-218/3 carrying human SOD1 genes were derived from the founder stock described by Epstein et al.(18). A linear 14.5-kb EcoRI-BamHI fragment containing the entire hCuZnSOD gene, was excised from the recombinant plasmid pHGSOD-SV neo and separated from plasmid sequences before microinjection. Approximately 500 copies of the purified fragment were microinjected into the male pronuclei. The founder mice had been bred with CD-1 mice to produce Tg offspring carrying the SOD1 gene. Tg mice were identified by qualitative demonstration of human CuZn/SOD, using nondenaturing gel electrophoresis followed by nitro blue tetrazolium staining as previously described(18). There were no observable phenotypic differences between Tg mice and nTg littermates.
Hypoxic-ischemic injury. We used a perinatal hypoxic-ischemic model developed by Rice et al.(1) that is based on the Levine(19) procedure(20). All animal research was approved by the University of California San Francisco Committee on Animal Research and was performed with the highest standards of humane care as set forth in the Guide for the Care and Use of Laboratory Animals, U.S. Department of Health and Human Services, Publication No. 85-23, 1985.
Briefly, 92 mice at postnatal d 7 were anesthetized with 2.5% halothane, 15% N2O, balance O2, and the right common carotid artery was exposed and occluded by electrical coagulation. The incision was sutured, and the pups were returned to their dams immediately after the surgery for at least 2 h to recover and feed. They were then placed in 1000-mL airtight containers partially submerged in a 37°C water bath through which a humidified atmosphere of 8% O2 and 92% N2 was introduced via inlet and outlet tubing. The pups remained in the hypoxic container for 90 min, and those that survived were returned to their dams.
Preparation and histologic scoring of brains. Five days after the hypoxic-ischemic procedure, the animals were anesthetized with 50 mg/kg pentobarbital intraperitoneally and perfused through the left ventricle with cold 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). The brains were immediately removed and postfixed in the same paraformaldehyde solution for 1-3 h and then transferred to cold 30% sucrose in 0.1 M phosphate buffer until sectioning. Coronal forebrain sections were cut at 50-μm intervals using a vibratome. Sections were stained with cresyl violet and then scored for degree of damage blindly as follows: 0, no detectable neuronal cell loss; 1, scattered neuronal cell loss with occasional astroglia; 2, columnar damage in the cortex involving predominantly layers II through IV; and 3, cystic infarction and gliosis.
Brain infarction. The extent of brain infarction was determined by measuring the area of surviving cortex with a video image analysis system using National Institutes of Health Image 1.47. The left and right cortices(contra- and ipsilateral) of three coronal sections from each brain were measured at the level of the anterior hippocampus by tracing the image which was calculated as mm2. Data are expressed as percent viable ipsilateral cortex over contralateral cortex, × 100.
Statistics. Mann-Whitney U test was used to analyze nonparametric ordinal data, and coded χ2 analysis was used for categorical data.
RESULTS
Survival data. Sixty-seven percent of the pups survived the procedure (Table 1). There was no difference in the survival of the Tg and nTg mice to the hypoxic-ischemic procedure (p= 0.602, χ2 test). Three Tg mice and three nTg mice who survived the procedure but were killed by their dams or died naturally before the time of sacrifice were considered to have survived the insult.
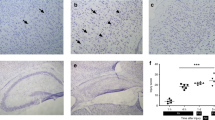
Histopathology. Figure 1 shows the damage that was observed in the Tg and nTg mice 5 d after the hypoxic-ischemic injury. Brain injury ranged from no detectable damage to extensive cystic infarction. The hippocampus was the most sensitive region to neuronal loss during hypoxia-ischemia in the mice regardless of the presence of the transgene. Only 17% of the Tg mice were free of histologic damage (score of 0), compared with 37% of the nTg mice. In addition, 48% of the Tg mice suffered cystic infarct (score of 3) compared with only 22% of the nTg mice(Table 2). Cystic infarctions varied in severity from destruction limited to portions of the cortex along the middle cerebral artery distribution to infarction that extended to the striatum and the thalamus, virtually destroying the affected hemisphere. Overall, the CuZn/SOD Tg mice had more severe histologic damage scores than their nTg litter-mates(p = 0.0463, Mann-Whitney U test).

Histopathologic damage by severity score. Coronal sections of mouse brain through the anterior hippocampus show increasing degrees of histopathologic damage by assigning a numerical value for the degree of damage observed under light microscopy. (A) Cresyl violet-stained section shows no neuronal loss (score 0); B, minimal neuronal cell loss and astroglia (arrow) (score 1); C, columnar infarction (score 2); D, cystic infarction and gliosis(score 3). Scale bar = 200 μm.
Brain infarction. In both Tg and nTg mice, there was a significant difference between the coronal area of the cortex contralateral to the common carotid artery occlusion and the area of remaining ipsilateral cortex. Because the contralateral area is assumed to be approximately equal to cortical area in nonischemic brains, the contralateral areas were used to express the degree of damage due to the hypoxia-ischemia. When areas are expressed as percentages (ipsilateral over contralateral) to minimize the effect of individual variation in brain size (Table 3), Tg mice had more extensive infarction than nTg mice (p = 0.0274, Mann-Whitney).
DISCUSSION
It was our goal to examine the role of superoxide radical formation in the pathogenesis of hypoxic-ischemic injury in 7-d-old mice. Our results demonstrate that CuZn/SOD Tg mice expressing three times normal amounts of CuZn/SOD suffered brain infarction more frequently than did nTg mice. Furthermore, the Tg mice had higher histologic severity scores than the nTg mice, indicating that they suffered more extensive brain damage. These findings suggest that brain injury in 7-d-old mice caused by hypoxia-ischemia is mediated in part by hydrogen peroxide production. The Tg animals did not die more frequently than the control littermates, suggesting that mortality in this model is related more to hemodynamic and cardiogenic compromise which was not evident in either group.
Hydrogen peroxide may account for the damage observed in our Tg mice by its accumulation in the brain due to elevated concentrations of the CuZn/SOD with normal levels of glutathione peroxidase. In the brains of uninjured nTg mice at postnatal d 7, CuZn/SOD mRNA and activity levels are reported to be four times greater than glutathione peroxidase levels(21). Although these activities are measurements obtained in vitro under pharmacologic, not physiologic, conditions, it is possible that this ratio may be increased under conditions of oxidative stress in vivo. The relative discrepancies in enzyme activities may result in an accumulation of hydrogen peroxide. This imbalance leading to hydrogen peroxide accumulation may account for some of the damage observed in the nTg mice. Increasing the activity of the CuZn/SOD, as in the Tg mice, without increasing GSH activity, would increase the accumulation of hydrogen peroxide and lead to greater damage. An accumulation of hydrogen peroxide may cause cellular damage by reacting with the superoxide radical in the presence of Fe2+ or Cu2+ to produce the hydroxyl radical, which is able to attack almost all cell constituents and damage cell membranes by initiating lipid peroxidation(22).
The above mechanism assumes that elevated levels of CuZn/SOD and/or hydrogen peroxide do not induce increased expression of GSH, leading to an increased detoxification of superoxide. Our results imply that induction does not occur, as the Tg mice exhibited more brain injury than the nTg mice instead of less injury as we would have expected if induction occurred. Glutathione concentrations are no different in the CuZn/SOD Tg mice than in nTg mice at P7 but during hypoxia-ischemia there is more consumption of GSH in Tg brains (Ferriero DM, Chan PH, unpublished observations). Similarly, Ceballos-Picot et al.(23) demonstrated that, at 2 mo, 12 mo, and 28 mo, glutathione peroxidase activity is not increased in Tg mice overexpressing CuZn/SOD when compared with the corresponding age group control. However, in the adult animals of the strain used in these experiments, glutathione peroxidase concentrations have been shown to rise in proportion to CuZn/SOD concentrations(24). In murine fibroblasts transfected with CuZn/SOD expression vectors, GSH peroxidase rises after exposure to the superoxide generator, paraquat(25). Further study is needed to determine whether CuZn/SOD and glutathione peroxidase are independently regulated during insults such as hypoxia-ischemia in the immature nervous system.
The outcome of ischemic injury may be different in the perinatal and adult brain due to changing proportions of superoxide detoxifying enzymes. When Chanet al.(26) subjected adults of this Tg strain to permanent middle cerebral artery occlusion, they found no differences between the Tg mice and their nTg littermates in infarct volume, local cerebral blood flow, or neurologic deficits. The difference between these results and our results suggests that enzymes responsible for the pathogenesis of brain injury may be differentially expressed in the perinatal and adult brain. Hothersall et al.(27) found that the activity of glutathione peroxidase was approximately 25% lower in uninjured 1-20-d-old rats compared with 4-mo-old rats, and about 40% lower compared with 12-mo-old rats. Glutathione reductase activity, on the other hand, rapidly increased between d 1 and 5, at which point the activity approximated the adult values(27). The exacerbation of damage observed in our 7-d-old mice compared with the lack of effect of CuZn/SOD overexpression in the adult mice of Chan et al. may have resulted from a decreased ratio of glutathione peroxidase to glutathione reductase in the perinatal mice.
There are many possibilities for the observed increased damage in the Tg brains. Another mechanism which may account for the increased damage observed in our Tg mice is increased toxicity from nitric oxide radical production. Nitric oxide radical (NO˙) is formed in the brain by NOS, a calcium/calmodulin-dependent enzyme. Calcium influx triggered byN-methyl-D-aspartic acid receptor activation during hypoxia-ischemia activates neuronal NOS, producing a significant local elevation in NO˙ production. This NO˙ may combine with the superoxide produced during hypoxia-ischemia to produce peroxynitrite. At physiologic pH, peroxynitrite degrades instantly to hydroxyl radical and nitrogen dioxide(NO2)(28). Although there is increased scavenging of superoxide in the CuZn/SOD Tg mice, which would decrease peroxynitrite, other mechanisms of nitric oxide-induced brain injury exist. There is an iron-independent reaction between NO˙ and hydrogen peroxide that leads to increase production of hydroxyl radical(29). In addition, at subphysiologic levels of intracellular L-arginine or tetrabiopterin, activation of brain NOS by calcium leads to formation of other reactive oxygen species instead of NO, especially hydrogen peroxide(30).
Further support of the hypothesis that NOS is involved in the pathogenesis of injury in these mice comes from the work of Oury et al.(31) who found that Tg adult mice overexpressing human extracellular SOD displayed increased sensitivity to CNS oxygen toxicity in an hyperbaric environment compared with nTg littermates. Inhibition of extracellular SOD and CuZn/SOD reduced the oxygen toxicity and more importantly, in the presence of a NOS inhibitor, the Tg mice no longer displayed any increased sensitivity to CNS oxygen toxicity compared with nTg littermates(31).
In summary, we have found that overexpression of SOD exacerbates perinatal hypoxic-ischemic brain injury. These data suggest that the neonatal brain may be unable to effectively detoxify hydrogen peroxide produced during periods of oxidative stress or that excess NO is produced in this setting. Further studies are underway to determine whether reactive oxygen species are critical determinants of injury after hypoxia-ischemia in the developing nervous system so that effective therapies aimed against their accumulation could prove beneficial.
Abbreviations
- Tg:
-
transgenic
- nTg:
-
nontransgenic
- SOD:
-
superoxide dismutase
- GSH:
-
reduced glutathione
- NOS:
-
nitric oxide synthase
References
Rice JE III, Vannucci RC, Brierley JB 1981 The influence of immaturity on hypoxic-ischemic brain damage in the rat. Ann Neurol 9: 131–141
Hill A 1991 Current concepts of hypoxic-ischemic injury in the term newborn. Pediatr Neurol 7: 317–325
Braughler JM, Hall ED 1989 Central nervous system trauma and stroke. In: Biochemical considerations for oxygen radical formation and lipid peroxidation. J Free Radic Biol Med 6: 289–301
Siesjo BK 1989 Free radicals and brain damage. Cerebrovasc Brain Metab Rev 1: 165–211
Carney JM, Starke-Reed PE, Oliver CN, Landum RW, Cheng MS, Wu JF, Floyd RA 1991 Reversal of age-related increase in brain protein oxidation decrease in enzyme activity, and loss in temporal and spatial memory by chronic administration of spin-trapping compoundN-tert-butyl-L-phenylnitrone. Proc Natl Acad Sci USA 88: 3633–3636
Chan PH, Kinouchi H, Epstein CJ, Carlson E, Chen SF, Imaizumi S, Yang GY 1993 Role of superoxide dismutase in ischemic brain injury: reduction of edema and infarction in transgenic mice following focal cerebral ischemia. Prog Brain Res 96: 97–104
Cathcart R, Schwiers E, Saul RL, Ames BN 1984 Thymine glycol and thymidine glycol in human and rat urine: a possible assay for oxidative DNA damage. Proc Natl Acad Sci USA 81: 5633–5637
Huang TT, Carlson EJ, Leadon SA, Epstein CJ 1992 Relationship of resistence to oxygen free radicals to CuZn superoxide dismutase activity in transgenic, transfected and trisomic cells. FASEB J 6: 903–910
Liu TH, Beckman JS, Freeman B, Hogan EL, Hsu CY 1989 Polyethylene glycol-conjugated superoxide dismutase and catalase reduce ischemic brain injury. Am J Physiol 256:H589–H593
He YY, Hsu CY, Ezrin AM, Miller MS 1993 Polyethylene glycol-conjugated superoxide dismutase in focal cerebral ischemia-perfusion. Am J Physiol 265:H252–H256
Chan PH, Longar S, Fishman RA 1987 Protective effects of liposome-entrapped superoxide dismutase on post-traumatic brain edema. Ann Neurol 21: 540–547
Turrens JF, Crapo JD, Freeman BA 1984 Protection against oxygen toxicity by intravenous injection of liposome entrapped catalase and superoxide dismutase. J Clin Invest 73: 87–95
Imaizumi S, Woolworth V, Fishman RA, Chan PH 1990 Liposome-entrapped superoxide dismutase reduces cerebral infarction in cerebral ischemia in rats. Stroke 21: 1312–1317
Chan PH, Kinouchi H, Epstein CJ, Carlson E, Chen SF, Imaizumi S, Yang GY 1993 Role of superoxide dismutase in ischemic brain injury: reduction of edema and infarction in transgenic mice following focal cerebral ischemia. Progr Brain Res 96: 97–104
Yang GY, Chan PH, Chen J, Carlson E, Chen SF, Weinstein P, Epstein CJ, Kamii H 1994 Human copper-zinc superoxide dismutase transgenic mice are highly resistant to reperfusion injury after focal cerebral ischemia. Stroke 25: 165–170
Palmer C, Vannucci RC, Towfighi J 1990 Reduction of perinatal hypoxic-ischemic brain damage with allopurinol. Pediatr Res 27: 332–336
Palmer C, Towfighi J, Roberts RL, Heitjan DF 1993 Allopurinol administered after inducing hypoxia-ischemia reduces brain injury in 7-day-old rats. Pediatr Res 33: 405–411
Epstein CJ, Avraham KB, Lovett M, Smith S, Elroy-Stein O, Rotman G, Bry C, Grooner Y 1987 Transgenic mice with increased Cu/Zn-superoxide dismutase activity: animal model of dosage effects in Down syndrome. Proc Natl Acad Sci USA 84: 8044–8048
Levine S 1960 Anoxic-ischemic encephalopathy in rats. Am J Pathol 36: 1–17
Ferriero DM, Arcavi LJ, Sagar SM, McIntosh TK, Simon RP 1988 Selective sparing of NADPH-diaphorase neurons in neonatal hypoxia-ischemia. Ann Neurol 24: 670–676
De Haan JB, Tymms MJ, Cristiano F, Kola I 1994 Expression of copper/zinc superoxide dismutase and glutathione peroxidase in organs of developing mouse embryos, fetuses, and neonates. Pediatr Res 35: 188–196
Moorhouse PC, Grootveld M, Halliwell B, Quinlan JG, Gutteridge JMC 1987 Allopurinol and oxypurinol are hydroxyl radical scavengers. FEBS Lett 213: 23–28
Ceballos-Picot I, Nicole A, Clement M, Bourre J, Sinet P 1992 Age-related changes in antioxidant enzymes and lipid peroxidation in brains of control and transgenic mice overexpressing copper-zinc superoxide dismutase. Mutat Res 275: 281–293
Przedborski S, Jackson-Lewis V, Kostic V, Carlson E, Epstein CJ, Cadet JL 1992 Superoxide dismutase, catalase, and glutathione peroxidase activities in copper/zinc-superoxide dismutase transgenic mice. J Neurochem 58: 1760–1767
Kelner MJ, Bagnell R 1990 Alteration of endogenous glutathione peroxidase, manganese superoxide dismutase, and glutathione transferase activity in cells transfected with a copper-zinc superoxide dismutase expression vector. J Biol Chem 265: 10872–10875
Chan PH, Kamii H, Yang G, Gafni J, Epstein CJ, Carlson E, Reola L 1993 Brain infarction is not reduced in SOD-1 transgenic mice after a permanent focal cerebral ischemia. NeuroReport 5: 293–296
Hothersall JS, El-Hassan A, McLean P, Greenbaum AL 1981 Age-related changes in enzymes of rat brain: redox systems linked to NADP and glutathione. Enzymes 26: 271–276
Chan PH 1994 Oxygen radicals in focal cerebral ischemia. Brain Pathol 4: 59–65
Halliwell B, Gutteridge JMC 1989 Free Radicals in Biology and Medicine. Oxford University Press, New York
Heinzel B, Mathias J, Klatt P, Bohme E, Mayer B 1992 Ca+/calmodulin-dependent formation of hydrogen peroxide by brain nitric oxide synthase. Biochem J 281: 627–630
Oury TD, Ho Y, Piantadosi CA, Crapo JD 1992 Extracellular superoxide dismutase, nitric oxide, and central nervous system O2 toxicity. Proc Natl Acad Sci USA 89: 9715–9719
Acknowledgements
The authors thank Elaine Carlson and Rhoda Gacayan for breeding the CuZn/SOD Tg mice and for transgene measurements, and Dr. Pak Chan for helpful discussions regarding the experiments and the manuscript.
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Ditelberg, J., Sheldon, R., Epstein, C. et al. Brain Injury after Perinatal Hypoxia-Ischemia Is Exacerbated in Copper/Zinc Superoxide Dismutase Transgenic Mice. Pediatr Res 39, 204–208 (1996). https://doi.org/10.1203/00006450-199602000-00003
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1203/00006450-199602000-00003
This article is cited by
-
Echinacoside Alleviates Hypoxic-Ischemic Brain Injury in Neonatal Rat by Enhancing Antioxidant Capacity and Inhibiting Apoptosis
Neurochemical Research (2019)
-
Pre-clinical models in pediatric traumatic brain injury—challenges and lessons learned
Child's Nervous System (2017)
-
MicroRNAs participate in the murine oligodendroglial response to perinatal hypoxia–ischemia
Pediatric Research (2014)
-
Glutathione peroxidase overexpression causes aberrant ERK activation in neonatal mouse cortex after hypoxic preconditioning
Pediatric Research (2012)
-
Necrostatin Decreases Oxidative Damage, Inflammation, and Injury after Neonatal HI
Journal of Cerebral Blood Flow & Metabolism (2011)
