
Abstract
Large cancer projects measure somatic mutations in thousands of samples, gradually assembling a catalog of recurring mutations in cancer. Many methods analyze these data jointly with auxiliary information with the aim of identifying subtype-specific results. Here, we show that somatic gene mutations alone can reliably and specifically predict cancer subtypes. Interpretation of the classifiers provides useful insights for several biomedical applications. We analyze the COSMIC database, which collects somatic mutations from The Cancer Genome Atlas (TCGA) as well as from many smaller scale studies. We use multi-label classification techniques and the Disease Ontology hierarchy in order to identify cancer subtype-specific biomarkers. Cancer subtype classifiers based on TCGA and the smaller studies have comparable performance, and the smaller studies add a substantial value in terms of validation, coverage of additional subtypes, and improved classification. The gene sets of the classifiers are used for threefold contribution. First, we refine the associations of genes to cancer subtypes and identify novel compelling candidate driver genes. Second, using our classifiers we successfully predict the primary site of metastatic samples. Third, we provide novel hypotheses regarding detection of subtype-specific synthetic lethality interactions. From the cancer research community perspective, our results suggest that curation efforts, such as COSMIC, have great added and complementary value even in the era of large international cancer projects.
Similar content being viewed by others

Introduction
Obtaining a comprehensive catalog of mutated genes in cancer is one of the holy grails of biomedical research. A catalog that links genes to cancer development and progression across many subtypes may enable accurate cancer diagnostics, and will form a basis for improving health care.1, 2 Large projects such as The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium (ICGC) have addressed the challenge by characterizing genomic data from thousands of patients across many cancer subtypes.3, 4, 5, 6, 7 The complexity of these data necessitated development of algorithms for delineating recurring mutations. For example, methods for detecting recurrence of single-nucleotide variations in genes were developed based on comparing the number of observed mutations in a gene to its background mutation rate. This is calculated across patients in order to distinguish between passenger and non-passenger events.8, 9
The results so far have been promising, yet recent studies showed that detection of pertinent mutated genes is still a hard task, mainly because most genes are mutated at intermediate frequencies (2–20%) or even lower.1, 2, 10 Additionally, different patients with the same cancer subtype tend to manifest low similarity in their mutation profiles.11 To cope with these problems, recently proposed methods made use of additional auxiliary information. For example, MutSigCV utilized gene expression data to better characterize the background mutation rate of short genes.1 HotNet used protein–protein interactions to detect connected subnetworks that harbor many mutated genes.10, 12 Hofree et al.11 stratified patients by quantifying the impact of their mutations on how information propagates in a protein–protein interaction network. ResponseNet and xseq modeled the impact of mutations on the gene expression profiles.13, 14 Liu et al.15 developed an ensemble method for detecting driver genes by integrating predictions from several approaches. Most of the analyses above reported cancer subtype-specific results. However, a thorough systematic assessment of the ability of somatic gene mutation profiles alone to predict the patient’s subtype is still missing. Furthermore, given the very large orchestrated data collection project, the utility and added value of smaller scale studies has been unclear.
To address those questions, we analyzed somatic mutation data of 9304 whole-exome samples from COSMIC, a database that contains somatic mutation profiles from the TCGA as well as many smaller studies.16 Each sample was represented by its mutated gene set, and a set of Disease Ontology (DO) terms that describe its phenotype.17 We applied on these data multi-label classification, where the goal is to construct an algorithm that predicts the set of labels (DO terms) for each sample based on its given features (the set of mutations observed in the sample). Such algorithm can also provide a set of specific biomarker genes for each disease.18 To the best of our knowledge this is the first application of multi-label classification to cancer mutation data. We tested a variety of multi-label classifiers using both leave-data sets-out cross-validation, and a stringent three-tier statistical validation process. In total, 20 out of 50 analyzed DO terms were well classified according to our validation, including bladder, pancreas, intestinal, leukemia, brain and benign neoplasms. A comparison of the TCGA and smaller studies showed that while the differences between the numbers of reported mutations are often immense, classifiers learned using the TCGA can predict subtypes in non-TCGA data, and vice versa.
For each cancer subtype our classifiers produce a set of relevant, subtype-specific signature genes. We demonstrate their value in three applications. First, these signatures recapitulate the main cancer genes in each subtype, and provide multiple novel candidate driver genes. Second, our classifiers can predict the primary site of a metastatic sample, suggesting potential clinical use for patients with cancer of unknown primary. Finally, we analyze our classifiers in the context of synthetic lethality (SL) interactions. SL interactions represent epistatic relations in which a double knockout of two genes results in a marked decrease in cell viability, whereas a single knockout of either gene is not lethal.19 These interactions were suggested previously as a basis for anticancer therapies.20, 21, 22 Here, we show a consistently significant over-enrichment of SL interactions between overmutated and under-mutated signature genes inferred from our classifiers. Thus, our analysis can propose novel subtype-specific candidate SL interactions between signature genes, with a promising therapeutic potential.
In summary, our key contributions are as follows: (1) construction and validation of disease-specific classifiers for 20 cancer types, (2) the first demonstration that mutations alone can successfully identify cancer types, (3) utilization of the resulting type-specific biomarker sets to discover novel driver genes, identify the origin of cancer of unknown primary, and pinpoint cancer type-specific SL interactions.
Results
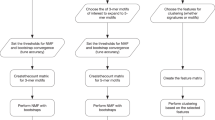
We first wished to test how well somatic mutation data from the COSMIC database can be used to classify cancer types, and whether there is added value in using information from small studies given the large international efforts. Our approach is summarized in Figure 1. We analyzed data sets from many studies covered by the COSMIC database. We merged these data sets by taking the high-quality binary gene–patient associations from whole-exome studies provided by COSMIC (see Materials and Methods). These data covered 9304 patients from 126 different studies. 4636 of the patients originated from TCGA studies. Genes (17 882) that had ⩾10 mutations across all patients were analyzed. We manually mapped each patient to its DO labels by using the restricted vocabulary of COSMIC for sample description. We used DO terms that had ⩾50 patients in ⩾3 different studies. We also removed general terms (for example, ‘cancer’). The DO terms are structured as a hierarchy where child–parent relations are of ‘is-a’ type (that is, the child is more specific). To avoid redundant terms, when a child and its parent had exactly the same patient set we removed the child. Overall, 50 DO terms remained.

Overview of the analysis. Samples and mutations were selected and annotated. Machine learning was used to identify cancer subtypes that could be reliably classified. For each well-classified subtype a gene signature was extracted from the classifier. The gene signature contains both over- and under-mutated genes. Finally, these sets are used for three applications: (1) gene-subtype associations, (2) primary site prediction given a metastatic sample and (3) novel predictions of subtype-specific synthetic lethality. A parallel effort compared the classification quality obtained on data from the small studies to that based on the TCGA information.
Improved subtype classification
We tested multi-label classification algorithms using 10-fold leave-data sets-out cross-validation.23, 24 That is, we excluded a tenth of the studies at a time while making sure that DO terms were always represented in the training set. We then learned classifiers using the training data sets and tested their performance on the excluded studies. As we observed previously,24 when analyzing a diverse collection of data sets spanning many diseases, performance evaluation requires several scores in order to avoid over-optimistic results. Given a disease term D, all samples are first partitioned to three groups: positives (P)—the samples with the disease D, negatives (N)—the samples that do not have D but originated from the same studies as the samples in P, and the background controls (BGC)—all others. To evaluate a classifier for D we calculated receiver-operating characteristic (ROC) scores for the separation between P and N (PN-ROC), as well as between P and BGC (PB-ROC). Area under the precision-recall curve measures were defined similarly (that is, PN- and PB-AUPR). We also calculated a meta-analysis q-value for the separation between P and N within studies.24 This test could be applied only for studies with a reasonable number of positives and negatives (we required at least five for each). Very few studies in our data satisfied this condition, and these usually covered only a small fraction of the set P. For example, while two astrocytoma studies satisfied this condition, they covered only 79 out of our 436 astrocytoma samples. We therefore performed the meta-analysis only if these studies covered ⩾5% of P. Twenty-two terms met these criteria.
We tested three types of classifiers: multi-label k nearest neighbors (MLkNN),25 HOMER26 and Binary Relevance18 (BR). Multi-label k nearest neighbors and HOMER consider dependencies among terms, whereas BR simply learns a separate binary classifier for each DO term. For BR we used feature selection to reduce running time (see Materials and Methods for details) and tested four classifiers: (1) support vector machines,27 (2) standard random forest,28 (3) Ranger—a fast implementation of random forest developed for genome-wide association studies29 and (4) Ranger DS—Ranger preceded by down-sampling the BGC and N populations. Except for Ranger DS all algorithms performed rather poorly (average ROC⩽0.6). For each classifier, term D was defined as well classified if the following conditions were satisfied:(1) PB-ROC⩾0.7; (2) if D had at least 10 negative samples, PN-ROC⩾0.7; and (3) if the meta-analysis could be applied we required q⩽0.1. The results are shown in Figure 2a. The top performing algorithm was Ranger DS with 20 well-classified terms, whereas the second best algorithm was Ranger with only 7 terms. Figure 2b shows that Ranger DS is superior both in ROC-PN and ROC-PB. We also tested Ranger with SMOTE, another method for sampling balanced data sets from imbalanced data.21, 30, 31 Supplementary Figure 1 shows a comparison of this method to Ranger DS. Although the two methods led to similar prediction quality, the SMOTE-based variant was much slower. Based on these results we used Ranger DS for all subsequent analyses.

Leave-datasets-out cross-validation. (a) For each classifier, the bars show (from bottom to top) the numbers of DO terms with PN-ROC ⩾7, PB-ROC ⩾0.7, and the number of well-classified terms. For binary relevance classifiers (BR) the number of features used is shown after the classifier name. (b) A comparison of Ranger and Ranger DS. Each point is a term.
Figure 3 shows the studied terms on the DO network. Well-classified terms include leukemia subtypes, brain cancer, liver cancer, intestinal cancer, urinary system cancer, benign neoplasm and others. In addition, some terms had high ROC scores and a marginal q-value (for example, mature B-cell neoplasm had PN-ROC=0.83, PB-ROC=0.82 and q=0.12). As a word of caution, out of the well-classified DO terms only four could be validated using the meta-analysis test. On the other hand, whenever the meta-analysis validation could be used, very few studies had both positive and negative samples. For example, for integumentary system cancer (PN-ROC=0.78, PB-ROC=0.77), out of 11 studies only one could be used and it contained only 15.7% of the positive samples. Larger community efforts are needed to provide heterogenous data sets that collect more than one cancer subtype.

The analyzed disease terms. Nodes are DO terms and edges reflect the DO hierarchy. Edges represent ‘is-a’ relations. Node size is proportional to the PB-ROC score, whereas the node color is proportional to the PN-ROC score. Nodes with wide borders mark well-classified terms.
Finally, we observed a significant correlation between the PN-ROC scores and the number of studies per DO term (Spearman r=0.4, P<0.01), and no correlation with the number of positive samples (Spearman r=0.004, P=0.9). Also, no additional well-classified terms can be gained by using the number of mutations in a patient as a single feature for classification. A possible explanation is the large differences between TCGA and non-TCGA studies; see the next section for a detailed discussion. In summary, our results show that although subtype classification based on mutation data is a hard task, high performance can be reached for many disease terms, with a clear bias towards those for which many studies were collected.
TCGA vs small studies
The 9304 samples analyzed above consist of two disjoint groups of roughly of the same size: TCGA (4636 samples, 17 studies) and non-TCGA samples, 4668 samples from 109 studies. The number of mutations per gene was highly correlated between the two groups (Figure 4a). However, when the number of mutations was counted per sample separately for each DO term, the differences between the groups were very high in most subtypes. For 22 out of 39 shared DO terms the difference between the distributions was significant (P<0.001, Bonferroni correction). In 10 cases the mutation frequency in the TCGA samples was lower. Figure 4b shows two examples in which the medians differed by more than 10-fold: integumentary system cancer and plasma cell neoplasm.

Comparison of 4636 TCGA samples to 4668 non-TCGA samples. (a) Number of mutations per gene. Each point gives the total number of mutations of a gene over all samples in each cohort. (b) Example of differences in the number of mutations in the two sets. In integumentary system cancer the TCGA samples have many more mutations. In plasma cell neoplasm the non-TCGA samples have more mutations. (c) Classification performance results: y-axis—AUC value when training on Non-TCGA and measuring the performance on the TCGA samples; x-axis—AUC values of the reverse test. Performance is measured by comparing positives vs the rest. Each point is a DO term. The difference between the two sets of AUC values is significant (P=0.003).
We also tested the ability of each group to predict the labels of the other. That is, we learn classifiers on the TCGA samples, and measure their performance on the non-TCGA samples, and vice versa. The results are shown in Figure 4c. We did not separate the non-positive cases into negatives and BGCs in these calculations because the TCGA studies had no negatives. Interestingly, using the non-TCGA samples for training produced better performance (P=0.003), with the majority of the tested terms achieving ROC>0.75. These results suggest that the cohorts share common local patterns that identify cancer subtypes, and that the non-TCGA samples provide substantial added value.
A simple scheme for interpreting a classifier
In the rest of the paper we show how our classifiers can provide insights on different cancer related problems. For a well-classified disease term D, we take the top 50 genes according to their importance score in the classifier, and call them the signature of D. Supplementary Figure 2 shows that using the top 50 genes likely covers the vast majority of the genes that were important for classification. For each signature gene we define its enrichment factor (EF) as the log ratio between the probabilities of observing a mutation in that gene in samples of disease D and in the rest of the samples. Supplementary Table 2 contains all the computed signatures and scores. For each cancer subtype D, a signature gene is called overmutated if EF>0 and under-mutated otherwise. Our overmutated gene sets recapitulate the main cancer genes in most examined cases, see Figure 5a for a comparison to Lawrence et al.1

Detected genes based on classifier interpretation. (a) A comparison of our signature genes to the gene sets detected by pan-cancer analysis of the TCGA data. Colored cells represent significant overlap (P<0.001), with color intensity showing the Jaccard coefficient between the two sets. (b–d) Subnetworks of signature genes. Edges are either protein–protein interactions or pathway interactions. Node size is proportional to the percent of mutated samples in the subtype. Node label size is proportional to the random forest importance score. Node color represents the gene enrichment factor (EF). Red: overmutated; Green: under-mutated. Blue: genes added by GeneMANIA. (b) Intestinal cancer. (c) Pancreatic cancer. (d) Organ system benign neoplasm. In b triangular nodes indicate genes overmutated according to TCGA colon cancer analysis. In c and d triangular nodes indicate genes that were identified as over-mutating in Pan-Cancer TCGA analysis.
Learning disease–gene associations
Figures 5b–d show network analysis examples for intestinal cancer, pancreatic cancer and benign neoplasm. The last two were not covered by Lawrence et al.1 and therefore could only be analyzed using COSMIC. The main connected component is shown in each case along with some additional high scoring genes. Many of the reported genes have a low mutation rate (for example,<5%, node size in the figure) and were not detected in Lawrence et al.1 For benign neoplasm the analysis captures some of the known cancer genes detected in the complete PanCan analysis. However, they are under-mutated (that is, less likely to be mutated in benign neoplasms compared with cancerous ones). Note that while this signature is very general and does not pertain to specific forms of benign neoplasm, it represents a general common denominator validated across all benign neoplasm studies in COSMIC.
Importantly, previous studies identified some highly mutated genes, such as TTN, MUC4 and MUC16, but then argued that they should be excluded based on biological relevance.8 In our analysis these genes were eliminated, appeared with a negative EF, or had lower importance scores (that is, were not in the top 50, or had lower importance scores compared with the genes in the network). Thus, our results both highlight important genes and ‘clean’ undesired effects. In addition, our results improve upon previous analyses based on the TCGA only. For example, in kidney cancer, TP53 has a negative EF score. That is, although it is marked as highly significantly mutated by the previous TCGA studies,1, 32 when adding the COSMIC samples its effect is reversed. Moreover, Lawrence et al.1 reported a set of candidate genes with marginal significance for each subtype, but very few of them had high good importance scores in our analysis. For example, in intestinal cancer only AXIN2 and FAM123B (AMER1) were detected.
In addition to eliminating genes with low importance scores, our analysis suggests novel, less frequently mutated candidates. Examples include FAM194B and IRF5 in intestinal cancer. LATS2 is another interesting candidate whose inactivation was shown to suppress P53 and inactivate cell migration.33 In addition, this gene is a known tumor suppressor that governs homeostasis.34 In pancreatic cancer, we discovered well known genes such as KRAS, SMAD4 and TP53.35, 36, 37, 38, 39, 40 We also discovered GLI3, a mediator of the hedgehog pathway activity, although it is mutated in only 4.1% of the patients. The hedgehog signaling pathway is responsible for maintaining pancreatic cancer stem cells, and thus it is a main candidate for treatment.41, 42 TGFBR2, a trans-membrane protein that binds TGF-beta, was discovered although it is mutated in only 3.7% of the patients. The TGF-beta signaling pathway is another major contributor to pancreatic cancer development.43 In addition, detection of GALR3, which is the receptor of the neuropeptide Galanin, may suggest that this pathway is important in pancreatic cancer and could lead to new insights on the pathogenesis and potential therapy.
Primary site prediction from a metastatic sample
Can the classifiers be used to identify the origin of cancer from metastatic samples? This would be highly valuable for cancers of unknown primary. We analyzed the data of Zhao et al.,44 which recently published exomes from 85 samples of different metastatic sites collected from 24 patients with a known primary site. Out of the 24, 13 patients had a primary site that could be mapped to at least one of our well-classified subtypes: 6 pancreatic cancer (21 metastatic samples), 1 intestinal cancer (2 samples), 2 kidney cancer (7 samples) and 4 female reproductive organ cancer (12 samples). Although the last subtype is very broadly defined, we included it in our analysis. Figure 6a shows the prediction results of our classifiers for each of the four subtypes on all 85 metastatic samples. For the three focused subtypes, the prediction quality, as measured by the separation between the metastatic samples from the correct primary site and the other metastatic samples was significant (P<0.02, ROC⩾0.85 in all cases). Thus, our classifiers learned from COSMIC successfully pointed out the correct primary sites for three different subtypes in an independent set of samples.

Primary cancer site prediction and synthetic lethality (SL) analysis. (a) The predictions of our classifiers on 85 metastatic samples from a new independent data set. Each boxplot shows the predictions of a specific classifier. In each subfigure, the left boxplot shows the probabilities assigned to metastatic samples whose known primary site fits that of the classifier, and the right shows the predictions on samples of other primary sites. (b) Enrichment analysis of SL interactions among the signature genes of each classifier. The barplots show the SL pair density within and between the over- and under-mutated signature gene sets (denoted as O and U, respectively). Only subtypes for which the number of edges between the sets was significant (q<0.1) are shown. (c) SL subnetworks in kidney and pancreatic cancers. Red nodes: overmutated genes, green nodes: under-mutated genes.
Enrichment of SL interactions
SL in cancer has recently drawn a lot of attention,19 and we reasoned that our analysis can expose a new facet of this phenomenon. For each well-classified cancer subtype, we partitioned the signature genes into two sets: O, the overmutated genes, and U, the under-mutated ones. As discussed above many under-mutated genes were detected in our analyses. We focused on the relations of the under- and overmutated genes for the same cancer subtypes. We reasoned that while accumulating mutations in the overmutated genes is likely to cover driver events that cause initialization and progression of cancer, similar mutations are not observed in the under-mutated gene set because that cells that randomly acquire these mutations do not survive. Hence, simultaneous mutations in an under-mutated gene and an overmutated gene would tend to be detrimental to the survival of the cancer cells. In other words, we expect high enrichment of SL interactions between genes in U and in O.
To test this reasoning, we analyzed the SynLethDB database of SL interactions in human.19 We measured the density of SL within set U, within set O and between the sets U and O, and also computed the significance of the number of O–U SL pairs observed (see Materials and Methods for details). Nine subtypes had significant results (Figure 6b). Interestingly, density is consistently lower within O genes, whereas in seven subtypes the O–U density was higher than O–O and U–U densities. Similar results were obtained when using the top 100 and 200 genes in terms of importance as the signature of the disease, see Supplementary Text. Figure 6c shows examples of the induced SL network of the gene sets in kidney and pancreatic cancers. In these networks, most SL interactions are between the under-mutated genes. These are highly connected to the overmutated genes, which are needed to keep the connectivity of the network intact.
Discussion
Advances in DNA sequencing technologies over the past decade have led to great progress in the endeavor of learning a catalog of mutated genes in cancer. Current databases provide data either from large projects such as the TCGA, or by curation efforts that span numerous smaller studies. Here, we highlighted the cumulative added value of smaller studies, as summarized by the COSMIC curation effort, in validating, refining, and extending the results produced by the large projects. Comparison of TCGA and non-TCGA studies showed that the differences between the numbers of reported mutations per subtype are high in most subtypes. On the other hand, classifiers learned using the TCGA can predict non-TCGA subtypes and vice versa. Interestingly, training the classifiers using the smaller studies was significantly better, which highlights the quality of these data.
Statistically, learning the associations between genes and cancer subtypes is difficult since most genes are mutated at low or intermediate frequencies.2 Unlike most methods, which incorporate additional data, we showed that classification can be done for many subtypes using somatic gene mutation data only. Notably, when projecting the genes used in the classifiers on interaction networks, connected subnetworks that contain many rarely mutated genes emerge. Interestingly, standard classification algorithms produced very low performance (Figure 2). Nevertheless, the top performing algorithm, which is based on Ranger with subsampling, gave high-quality predictions in 20 out of 50 tested DO terms. Validating a classifier required using leave-data sets-out cross-validation and combining three different performance criteria. As few studies include samples of several different cancer subtypes, some of our criteria could not always be calculated. Future studies that mix several subtypes will likely achieve better results.
The number of data sets available for each disease is a key factor in classification success. We observed a positive correlation between classification quality and the number of data sets. Terms with a small number of data sets tend to have fewer positive and negative samples as well as lower biological heterogeneity (as different studies often cover distinct populations). Other factors may impair classification quality: some disease term definitions may be too broad (for example, endocrine gland cancer, which had >1,000 samples, achieved PB-ROC=0.62). Also, when the sample set of a disease is too similar to that of a parent or a child subtype in the DO hierarchy, the ROC score for separating between them may be low (as can be seen, for example, for the colorectal cancer branch in Figure 3). Finally, lack of discriminating somatic mutations between similar terms can result in weak separation between them.
We have shown how a very simple analysis based on the gene importance scores obtained from the classifiers can recapitulate, refine and extend the recent results from the large projects. Unlike other approaches like TumorPortal,1 which highlights genes for a particular cancer type based on their mutation rate only, our approach prefers genes that are specific to the type, and avoids genes that are non-specific even if they have high mutation rate. For example, TP53 in liver cancer is mutated in >20% of the samples, which is 1.2-fold lower than in other cancer types. This gene was marked as down-represented in liver cancer in our analysis (Supplementary Table 1).
Although our results are promising, they have several limitations. The first and foremost is data availability: we could only cover to terms with at least three disjoint studies in order to perform a rigorous validation of our multi-label classification flow. On the other hand, some of the resulting DO terms correspond to very general cancer types, with no direct clinical usage. Nevertheless, as shown in Figure 5a, many of our well-classified DO terms are similar to terms defined and analyzed by the TCGA, which marks the state of the art of large-scale pan-cancer analysis. Currently we used the top 50 genes based on their importance, as was suggested previously,15 and do not assign significance scores to single genes. Future methods that will integrate the gene importance score within a significance testing model are expected to be more powerful in detecting novel genes. The approach is powerful for some cancer types, but does not provide consistent result according to all three criteria in others, like breast and ovarian cancer. This could be due to the heterogeneity of these tumors, or due to the inability to distinguish a class from its subclass.
Another limitation is that we only used binary somatic mutation profiles and ignored additional information on the quantity and quality of the mutations (for example, zygosity information). Our analysis is also limited by the decisions made by the data curators (for example, selection bias towards some subtypes), the resolution of the DO hierarchy, and in considering only mutations in exons.
We demonstrated the utility of our analysis for three applications, but these should be viewed as proofs of concept that require further development. While we could predict the primary site of a metastatic sample significantly and with high ROC scores, the sample size used in this analysis was rather small; additional studies using more samples are required to establish the usability of our classifiers in practice. Our SL analysis detected enrichment of SL interactions between the over- and under-mutated gene sets for most well-classified DO terms. These results corroborate our hypothesis that the observed cancer cells are those that tended to avoid having mutations in the under-mutated gene set. However, further research is required for inference of specific pair-wise interactions. Finally, using our network summarizations (Figure 4b) we were able to point out connected subnetworks related to functional pathways (for example, hedgehog signaling in pancreatic cancer), which contained rarely mutated genes. Detection of such subnetworks is of high interest as most driver genes are expected to be rarely mutated.10 Such modules can also be used to suggest new hypotheses, but these must be tested experimentally.
Materials and methods
COSMIC data
We downloaded the complete gene somatic mutation data table from COSMIC (February 2015 version), where rows describe a single mutation in a specific patient. We kept only rows that were: (1) ‘Confirmed somatic variant’ (that is, the mutation passed the analysis of the COSMIC curators) and (2) originated from ‘Genome-wide screen’. This filter resulted in 1 393 387 surviving gene–patient pairs. In 87.5% of those the gene was mutated only once in that patient, and for 92.5% of the mutations zygosity information was not available. These numbers suggest that our decision to use binary gene–patient relations does not cause a major loss of information.
We used COSMIC’s study ids to partition the samples by their studies. When no COSMIC id was available we used Pubmed ID. This resulted in 221 studies. We then repeatedly united studies that overlapped in their samples, until we were left with 126 patient-disjoint studies. We merged the data from the different studies by forming a gene–patient binary matrix recording which genes had one or more somatic mutations in each patient. Finally, we excluded 1369 genes that were mutated in <10 patients, leaving us with 17 882 genes.
Classification algorithms
We used the Mulan package for multi-label classification algorithms.45 For binary classification we used standard R implementation of linear support vector machines,27 random forest with 500 trees28 and Ranger29 with 1000 trees. For all binary classifiers we used feature selection before learning the classifiers in order to reduce running time. Here we selected the top 250 over-represented and top 250 under-represented genes using Fisher’s exact test. For support vector machines we selected the top 100 over-represented and top 100 under-represented genes, since for some DO terms the learning process did not converge in a reasonable running time (>4 h).
Ranger DS
Given disease term D with a set of patients P we created a collection of M=50 random forests as follows. For each i=1,…,M, we randomly selected |P| samples out of the non-D samples and called the resulting set negative sample set Ni. We then learned a Ranger classifier with 20 trees using P and Ni. Prediction on a new sample is done by reporting the average prediction over the 50 random forests.
Network visualization and analysis
Network visualization and analysis were done in Cytoscape46 using GeneMania.47, 48
Preprocessing the metastases data
We downloaded the Supplementary Data of Zhao et al.44 These data contained all somatic point mutations for each sample (we used the 85 metastatic samples of patients with a known primary site). We then mapped these positions to the GRCh37 genomic positions using the biomaRt R package.49 For each gene in our original training data, we checked in each of the samples whether at least one of its point mutations fell in the gene’s genomic position. This process created a somatic mutation binary profile for each metastatic sample, which was later used as a test set for our classifiers.
Synthetic lethality analysis
We defined the set O of overmutated genes as those with EF>0, and the set U of under-mutated genes as those with EF<0. For the SL network we used all SL gene pairs from SynLethDB.19
Significance estimation of the edges between two gene sets
For each well-classified subtype we analyzed the density of O–O, U–U and O–U pairs In the SL network G=(V, E). We removed genes that are not in V from O and U. For each node v in O we estimated the significance of the number of edges between v and U using the hyper-geometric test as proposed previously.50, 51 Here, the number of successful draws is x, the number of draws is the degree of v in G, the population size is |V|−1, and the size of the subpopulation on which success is measured is |U|. Finally, we merged the P-values obtained for each node in O using Fisher’s meta-analysis test. Our analysis can be viewed as a statistical test for the connectivity between O and U, conditioned on the degrees of the nodes in O.
Code availability
R implementation of the multi-label classifiers and the validation methods can be freely obtained for academic use at http://acgt.cs.tau.ac.il/adeptus/
References
Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway L a, Golub TR et al. Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 2014; 505: 495–501.
Raphael BJ, Dobson JR, Oesper L, Vandin F . Identifying driver mutations in sequenced cancer genomes: computational approaches to enable precision medicine. Genome Med 2014; 6: 5.
Cancer Genome Atlas Research Network. Comprehensive genomic characterization of squamous cell lung cancers. Nature 2012; 489: 519–525.
The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012; 490: 61–70.
The International Cancer Genome Consortium. International network of cancer genome projects. Nature 2010; 464: 993–998.
The International Cancer Genome Consortium. Computational approaches to identify functional genetic variants in cancer genomes. Nat Methods 2013; 10: 723–729.
Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K et al. The cancer genome atlas pan-cancer analysis project. Nat Genet 2013; 45: 1113–1120.
Lawrence MS, Stojanov P, Polak P, Kryukov G V, Cibulskis K, Sivachenko A et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013; 499: 214–218.
Dees ND, Zhang Q, Kandoth C, Wendl MC, Schierding W, Koboldt DC et al. MuSiC: identifying mutational significance in cancer genomes. Genome Res 2012; 22: 1589–1598.
Leiserson MDM, Vandin F, Wu H-T, Dobson JR, Eldridge J V, Thomas JL et al. Pan-cancer network analysis identifies combinations of rare somatic mutations across pathways and protein complexes. Nat Genet 2014; 47: 106–114.
Hofree M, Shen JP, Carter H, Gross A, Ideker T . Network-based stratification of tumor mutations. Nat Methods 2013; 10: 1108–1115.
Vandin F, Clay P, Upfal E, Raphael BJ . Discovery of mutated subnetworks associated with clinical data in cancer. Pac Symp Biocomput 2012. 55–66.
Ding J, McConechy MK, Horlings HM, Ha G, Chun Chan F, Funnell T et al. Systematic analysis of somatic mutations impacting gene expression in 12 tumour types. Nat Commun 2015; 6: 8554.
Lan A, Smoly IY, Rapaport G, Lindquist S, Fraenkel E, Yeger-Lotem E . ResponseNet: revealing signaling and regulatory networks linking genetic and transcriptomic screening data. Nucleic Acids Res 2011; 39: W424–W429.
Liu Y, Tian F, Hu Z, DeLisi C . Evaluation and integration of cancer gene classifiers: identification and ranking of plausible drivers. Sci Rep 2015; 5: 10204.
Forbes SA, Beare D, Gunasekaran P, Leung K, Bindal N, Boutselakis H et al. COSMIC: exploring the world’s knowledge of somatic mutations in human cancer. Nucleic Acids Res 2014; 43: D805–D811.
Schriml LM, Arze C, Nadendla S, Chang Y-WW, Mazaitis M, Felix V et al. Disease ontology: a backbone for disease semantic integration. Nucleic Acids Res 2012; 40: D940–D946.
Zhang ML, Zhou ZH . A review on multi-label learning algorithms. IEEE Trans. Knowl. Data Eng 2014; 26: 1819–1837.
Guo J, Liu H, Zheng J . SynLethDB: synthetic lethality database toward discovery of selective and sensitive anticancer drug targets. Nucleic Acids Res 2015; 44: D1011–D1017.
Whitehurst AW, Bodemann BO, Cardenas J, Ferguson D, Girard L, Peyton M et al. Synthetic lethal screen identification of chemosensitizer loci in cancer cells. Nature 2007; 446: 815–819.
Turner NC, Lord CJ, Iorns E, Brough R, Swift S, Elliott R et al. A synthetic lethal siRNA screen identifying genes mediating sensitivity to a PARP inhibitor. EMBO J 2008; 27: 1368–1377.
Jerby-Arnon L, Pfetzer N, Waldman YY, McGarry L, James D, Shanks E et al. Predicting cancer-specific vulnerability via data-driven detection of synthetic lethality. Cell 2014; 158: 1199–1209.
Lee YS, Krishnan A, Zhu Q, Troyanskaya OG . Ontology-aware classification of tissue and cell-type signals in gene expression profiles across platforms and technologies. Bioinformatics 2013; 29: 3036–3044.
Amar D, Hait T, Izraeli S, Shamir R . Integrated analysis of numerous heterogeneous gene expression profiles for detecting robust disease-specific biomarkers and proposing drug targets. Nucleic Acids Res 2015; 43: 7779–7789.
Zhang ML, Zhou ZH . ML-KNN: a lazy learning approach to multi-label learning. Pattern Recognit 2007; 40: 2038–2048.
Tsoumakas G, Katakis I, Vlahavas I . Effective and efficient multilabel classification in domains with large number of labels. Proceedings of the ECML/PKDD 2008 Workshop on Mining Multidimensional Data. 2008;30-44.
Cortes C, Vapnik V . Support vector machine. Mach Learn 1995. 1303–1308.
Breiman L . Random forests. Mach Learn 2001; 45: 5–32.
Wright MN, Ziegler A . ranger: a fast implementation of random forests for high dimensional data in C++ and R. 2015. Available at: https://arxiv.org/abs/1508.04409.
Chawla N V, Bowyer KW, Hall LO, Kegelmeyer WP . SMOTE: synthetic minority over-sampling technique. J Artif Intell Res 2002; 16: 321–357.
Torgo L . Data Mining With R - Learning With Case Studies. CRC Press, 2011, page 289.
The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of clear cell renal cell carcinoma. Nature 2013; 499: 43–49.
Furth N, Ben-Moshe NB, Pozniak Y, Porat Z, Geiger T, Domany E et al. Down-regulation of LATS kinases alters p53 to promote cell migration. Genes Dev 2015; 29: 2325–2330.
Visser S, Yang X . LATS tumor suppressor: a new governor of cellular homeostasis. Cell Cycle 2010; 9: 3892–3903.
Eser S, Schnieke A, Schneider G, Saur D . Oncogenic KRAS signalling in pancreatic cancer. Br J Cancer 2014; 111: 1–6.
Morris JP, Wang SC, Hebrok M . KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat Rev Cancer 2010; 10: 683–695.
Ji Z, Mei FC, Xie J, Cheng X . Oncogenic KRAS activates hedgehog signaling pathway in pancreatic cancer cells. J Biol Chem 2007; 282: 14048–14055.
Tascilar M, Skinner HG, Rosty C, Sohn T, Wilentz RE, Offerhaus GJA et al. The SMAD4 protein and prognosis of pancreatic ductal adenocarcinoma. Clin Cancer Res 2001; 7: 4115–4121.
Bardeesy N, Cheng KH, Berger JH, Chu GC, Pahler J, Olson P et al. Smad4 is dispensable for normal pancreas development yet critical in progression and tumor biology of pancreas cancer. Genes Dev 2006; 20: 3130–3146.
Maitra A, Hruban RH . Pancreatic cancer. Annu Rev Pathol 2008; 3: 157–188.
Onishi H . Hedgehog signaling pathway as a new therapeutic target in pancreatic cancer. World J Gastroenterol 2014; 20: 2335.
Kelleher FC . Hedgehog signaling and therapeutics in pancreatic cancer. Carcinogenesis 2011; 32: 445–451.
Truty MJ, Urrutia R . Basics of TGF-beta and pancreatic cancer. Pancreatology 2007; 7: 423–435.
Zhao ZM, Zhao B, Bai Y, Iamarino A, Gaffney SG, Schlessinger J et al. Early and multiple origins of metastatic lineages within primary tumors. Proc Natl Acad Sci USA 2016; 113: 2140–2145.
Tsoumakas G, Spyromitros-Xioufis E, Vilcek J, Vlahavas I . MULAN: a Java library for multi-label learning. J Mach Learn Res 2011; 12: 2411–2414.
Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D et al. Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 2003; 13: 2498–2504.
Montojo J, Zuberi K, Rodriguez H, Kazi F, Wright G, Donaldson SL et al. GeneMANIA cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics 2010; 26: 2927–2928.
Vlasblom J, Zuberi K, Rodriguez H, Arnold R, Gagarinova A, Deineko V et al. Novel function discovery with GeneMANIA: a new integrated resource for gene function prediction in Escherichia coli. Bioinformatics 2014. 1–5.
Durinck S, Moreau Y, Kasprzyk A, Davis S, De Moor B, Brazma A et al. BioMart and Bioconductor: a powerful link between biological databases and microarray data analysis. Bioinformatics 2005; 21: 3439–3440.
Ulitsky I, Shamir R . Identification of functional modules using network topology and high-throughput data. BMC. Syst Biol 2007; 1: 8.
Amar D, Shamir R . Constructing module maps for integrated analysis of heterogeneous biological networks. Nucleic Acids Res 2014; 42: 4208–4219.
Acknowledgements
This study was supported in part by the Israel Science Foundation (grant 317/13), an IDEA grant from the Dotan Center in Hemato-Oncology, and the Israeli Center of Research Excellence (I-CORE), Gene Regulation in Complex Human Disease, Center No 41/11. DA is grateful to the Azrieli Foundation for the award of an Azrieli Fellowship. DA was also supported in part by fellowships from the Edmond J. Safra Center for Bioinformatics at Tel Aviv University. Part of the work was done while DA and RS were visiting the Simons Institute for the Theory of Computing.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies this paper on the Oncogene website
Rights and permissions
This work is licensed under a Creative Commons Attribution-NonCommercial-ShareAlike 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-sa/4.0/
About this article

Cite this article
Amar, D., Izraeli, S. & Shamir, R. Utilizing somatic mutation data from numerous studies for cancer research: proof of concept and applications. Oncogene 36, 3375–3383 (2017). https://doi.org/10.1038/onc.2016.489
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2016.489
This article is cited by
-
Predicting links between tumor samples and genes using 2-Layered graph based diffusion approach
BMC Bioinformatics (2019)
-
Statistical representation models for mutation information within genomic data
BMC Bioinformatics (2019)
-
Predicting cancer type from tumour DNA signatures
Genome Medicine (2017)
