
Abstract
RASSF1A is a key tumor-suppressor gene that is often inactivated in a wide variety of solid tumors. Studies have illustrated that RASSF1A plays vital roles in the regulation of cell-cycle progression and functions as a guardian of mitosis. Nevertheless, the precise mechanism of RASSF1A-dependent regulation of mitosis remains largely unclear. APC/CCdc20 is the master switch and regulator of mitosis. The activity of APC/CCdc20 is tightly controlled by phosphorylation and specific inhibitors to ensure the sequential ubiquitination of downstream targets. Here, we report on the novel finding of a regulated circuitry that controls the timely expression and hence activity of APC/CCdc20 during mitosis. Our study showed that RASSF1A and APC/CCdc20 form a molecular relay that regulates the APC/CCdc20 activity at early mitosis. We found that RASSF1A inhibits APC/CCdc20 function through its D-box motifs. Paradoxically, RASSF1A was also demonstrated to be ubiquitinated by APC/CCdc20 in vitro and degraded at prometaphase despite of active spindle checkpoint presence. The first two unique D-boxes at the N-terminal of RASSF1A served as specific degron recognized by APC/CCdc20. Importantly, we found that Aurora A and Aurora B directly phosphorylate RASSF1A, a critical step by which RASSF1A switches from being an inhibitor to a substrate of APC/CCdc20 during the course of mitotic progression. As a result of RASSF1A degradation, APC/CCdc20 can then partially activate the ubiquitination of Cyclin A in the presence of spindle checkpoint. This circuitry is essential for the timely degradation of Cyclin A. To conclude, our results propose a new model for RASSF1A–APC/CCdc20 interaction in ensuring the sequential progression of mitosis.
Similar content being viewed by others


Introduction
RASSF1A is a potent tumor suppressor that is known to induce cell-cycle arrest. RASSF1A has been shown to be a mitotic regulator and it regulates G2/M transition by sequestering Cdc20 from APC/C, and hyperstabilizing the microtubules (Rong et al., 2004; Song et al., 2004). Aurora A and B are mitotic kinases that regulate the mitotic progression (Carmena et al., 2009). Recent studies revealed that RASSF1A is a substrate of Aurora A and the action of RASSF1A is regulated by Aurora A phosphorylation (Rong et al., 2007; Song et al., 2009). Particularly, Rong et al. (2007) showed that RASSF1A can no longer hyperstabilize microtubules after being phosphorylated by Aurora A. On the other hand, Song et al. (2009) suggested that Aurora phosphorylation induces RASSF1A-Cdc20 dissociation. However, the precise mechanism of RASSF1A on G2/M regulation remains elusive.
Anaphase-promoting complex/cyclosome (APC/C) is a ubiquitin ligase that ubiquitinates mitotic proteins and directs them to proteasomal degradation (Peters, 2006). APC/C is sequentially activated by Cdc20 and Cdh1 during mitosis. In early mitosis, Cdc20 accumulates and activates the catalytic activity of APC/C in the ubiquitination of mitotic cyclins and securin via recognition of D-box domain (King et al., 1996). Cdh1 later takes over from Cdc20 as the APC/C activator from anaphase to G1 phase, and the APC/CCdh1 recruits target substrates containing either D-box or KEN-box (Pfleger and Kirschner, 2000).
The activity of APC/C is sequentially regulated. During S phase and early mitosis, the activity of APC/CCdc20 is regulated by Emi1 and mitotic checkpoint complex at the spindle assembly checkpoint (SAC) (Miller et al., 2006; Peters, 2006). Once all kinetochores are attached to the mitotic spindle and the pulling force is established, SAC is turned off and APC/CCdc20 is fully activated to ubiquitinate securin. This leads to the consequential activation of separase and the initiation of anaphase (Pines, 2006).
It is suggested that APC/CCdc20 ubiquitinates its substrate in a SAC-dependent or SAC-independent manner. In the presence of SAC, APC/CCdc20 ubiquitinates Cyclin A and NEK2A, but not Cyclin B and Securin, despite that APC/CCdc20 recognized the D-boxes of these proteins as degron (Geley et al., 2001; Hames et al., 2001; Zur and Brandeis, 2001; Yamano et al., 2004). This discrepancy can be explained by the conformational change of substrate-recognition pocket of APC/CCdc20 induced by SAC (Izawa and Pines, 2011). APC/CCdc20-dependent ubiquitination is likely to be controlled by multiple factors. It was shown that binding between Cyclin A and Cks was essential for the onset of SAC-independent degradation of Cyclin A (Wolthuis et al., 2008). Importantly, expression of nondegradable Cyclin A induced mitotic delay (Geley et al., 2001; den Elzen and Pines, 2001). Degradation of NEK2A is necessary for the re-establishment of intercentriolar linkage of the centrosome cycle (Hames et al., 2001). These examples highlighted the importance of degrading the mitotic proteins by APC/C on time during mitotic progression.
Here, we discovered a regulatory mechanism by which the expression of RASSF1A could be precisely controlled during mitosis. Our findings provide explanations for the physiological function of RASSF1A in the timely entry and exit of mitotic cell division. We suggest that RASSF1A acts as an inhibitor of APC/CCdc20 through its D-box domains at early mitosis, but becomes a substrate of APC/CCdc20 as mitosis progresses. This transformation was demonstrated to be controlled through Aurora-dependent phosphorylation of RASSF1A. As mitosis progresses, the clearance of RASSF1A inhibition leads to reactivation of APC/CCdc20, which permits APC/CCdc20 to ubiquitinate Cyclin A, and launches the subsequent course of mitosis to late anaphase. This mechanism ensures a timely degradation of Cyclin A and NEK2A during mitosis progression.
Results
RASSF1A is degradable by APC/CCdc20
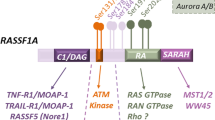
Structural analysis revealed that RASSF1A protein has six consensus D-boxes and one KEN-box (Figures 1a and b). As both D-box and KEN-box are peptide domains well recognized as APC/C degrons in directing proteasomal degradation, it is plausible that RASSF1A is a substrate of APC/C. We therefore determined whether RASSF1A is degradable in APC/C-dependent manner during mitosis. First, the protein level of RASSF1A was traced in HeLa cells synchronized by double thymidine block and release (Figure 2a). Our result showed that RASSF1A degraded at 8 h post-thymidine release. Both fluorescence-activated cell sorting (FACS) analysis and degradation of Cyclin A suggested that the HeLa cells entered G2/M phase at 8 h post-thymidine release, concomitant to the activation of APC/CCdc20 (Geley et al., 2001). Next, we asked whether the degradation of RASSF1A depends on spindle checkpoint. To address this question, HeLa cells synchronized by double thymidine block were released into medium containing nocodazole to activate the spindle checkpoint. Cell cycle was traced by immunoblotting of Cyclin A and Cyclin B1 and FACS analysis. As expected, Cyclin B1, Cdc20, Aurora A and Aurora B accumulated with time following the release from thymidine block (Figure 2b, left panels). The level of RASSF1A remained steady for the first 8 h, after which the protein level of RASSF1A and Cyclin A both dropped with time. This phenomenon proceeded independent of the nocodazole-induced spindle arrest. To determine the protein level of RASSF1A during mitotic exit, mitotic HeLa cells were released into medium without nocodazole. Progressive degradation of Cyclin B1, Cdc20, Aurora A and Aurora B implied that cells had completed anaphase and were en route for mitotic exit (Figure 2b, right panel). The level of RASSF1A rose after 4 h post-nocodazole release when the lowest level of Cdc20 was detected. Concomitantly, FACS analysis showed that the cells entered G1 phase after 4 h post-nocodazole release. The data indicated that RASSF1A was degraded during mitosis and accumulated at G1 phase. Taken together, we suggested that RASSF1A is a putative substrate of APC/C and is degraded at early mitosis.

RASSF1A contains multiple D-boxes and KEN-box. (a) Alignment of RASSF1A and RASSF1C amino-acid sequences. The putative D-boxes and KEN-box are highlighted in blue and red, respectively. The reported Aurora A phosphorylation site is highlighted in pink. (b) Schematic diagram of multiple putative D-boxes and one KEN-box present on RASSF1A. The sequences of the putative D-boxes and KEN-box are shown. * denotes every tenth amino acid in the amino acid sequence.

Cdc20 induces RASSF1A degradation. The protein level of RASSF1A fluctuates across the cell cycle progression. HeLa cells were synchronized by double thymidine block followed by releasing into medium without thymidine (a), double thymidine block followed by releasing into nocodazole (b, left panel), or nocodazole block and release (b, right panel). Cells collected at indicated time points were analyzed by immunoblotting. Similar to Cyclin A, RASSF1A was degraded at 8 h after releasing from thymidine block in the presence or absence of nocodazole. (c) Ectopic expression of Cdc20 downregulates RASSF1A. HeLa cells were transfected with vectors encoding FLAG-Cdc20(7A) or FLAG-Cdh1 and synchronized by double thymidine block, followed by releasing into nocodazole. The level of RASSF1A at indicated time points were analyzed by immunoblotting. (d) Knocking down of Cdc20 by siRNA stabilizes RASSF1A during early mitosis. HeLa cells transfected with indicated siRNA were synchronized and analyzed as in (c).
The possible mechanisms behind the mitotic degradation of RASSF1A were then examined. There are two activators of APC/C: Cdc20 and Cdh1. Using HeLa cells transfected with Cdc20 or Cdh1 and synchronized by double thymidine block, we determined whether both Cdc20 and Cdh1-activated APC/C induced the degradation of RASSF1A. The protein level of RASSF1A was analyzed by immunoblotting. Whereas overexpression of the active mutant of Cdc20 (Cdc20(7A)) promoted the premature downregulation of RASSF1A, Cdh1 did not induce any effect on RASSF1A (Figure 2c). Consistently, downregulation of RASSF1A was observed in Cdc20-overexpressed HeLa cells but not in Cdh1-overexpressed cells (Supplementary Figure 1). Furthermore, the Cdc20-dependent downregulation of RASSF1A was rescued by the treatment of proteasome inhibitor MG-132 (Supplementary Figure 1). Cdc20 may induce RASSF1A degradation via proteasome. The Cdc20-dependent degradation of RASSF1A was also confirmed in small interfering RNA (siRNA)-transfected cells. HeLa cells were transfected with siRNA targeting Cdc20 or Cdh1 and synchronized by double thymidine block and release (Figure 2d). During mitosis, RASSF1A was degraded in cells transfected with Cdh1 siRNA. This was similar to the cells transfected with control siRNA. However, RASSF1A was stabilized in cells transfected with Cdc20 siRNA. Our data thus suggested that Cdc20 induced proteasome-dependent degradation of RASSF1A at early mitosis.
Cdh1 and Cdc20 bind to the substrate of APC/C and target them to ubiquitination (Pfleger and Kirschner, 2000; Yu, 2007). Thus, we further determined the interaction between endogenous RASSF1A and Cdc20 by immunoprecipitation assay. We found that RASSF1A interacted with Cdc20 in mitotic HeLa cells and mitotic HeLa cells treated with proteasome inhibitor MG-132 (Figure 3a). On the contrary, our immunoprecipitation assay did not suggest any interaction between Cdh1 and RASSF1A in asynchrony or mitosis. The finding indicated that Cdc20 is capable of binding to RASSF1A preferentially in mitotic cells. To define the specific Cdc20-interacting domains on RASSF1A, immunoprecipitation was performed on cells co-transfected with Cdc20 and deletion mutants of RASSF1A. However, we found that both N-terminus and C-terminus deletion constructs of RASSF1A maintained immunoprecipitating ability with Cdc20 (Figure 3b). As there are multiple D-boxes and a KEN-box on RASSF1A, we performed motif mapping assay to examine the interaction, if any, between these specific RASSF1A domains and APC/C. Subsequent constructs made with specific D-boxes (DBD) and KEN-box (KD) deleted also did not seem to alter the binding affinity between RASSF1A and Cdc20 (Figure 3c). To further delineate the Cdc20-binding domain on the N-terminus and C-terminus of RASSF1A, immunoprecipitation was done on cells co-transfected with Cdc20 and truncated RASSF1A without D-boxes and KEN-box (Figure 3d). Concurring with Figure 3c, full-length RASSF1ADBD1−6+KD bound to Cdc20. However, the N-terminus of RASSF1A with the D-boxes being deleted failed to bind to Cdc20 (1–140DBD+KD). This interesting finding suggested that the two D-boxes at the N-terminus of RASSF1A were Cdc20-binding domains. It is concordant with the hypothesis that Cdc20 targets D-boxes in recruiting substrates to APC/C (Yu, 2007). Intriguingly, the Cdc20-binding ability was retained in 1–264DBD+KD and 141–340DBD+KD RASSF1A mutants. It is suggested that there is D-box-independent Cdc20-binding domain on the amino acids 141–264 of RASSF1A. Our findings indicated that there are D-box-dependent and D-box-independent Cdc20-interacting domains on RASSF1A.

RASSF1A interacts with Cdc20 during mitosis. (a) RASSF1A was immunoprecipitated from asynchronized (AS), mitotic (M) or MG132-treated mitotic (M+MG132) HeLa cells as indicated. The immunoprecipitate and 2% of the input WCL was subjected to immunoblotting (IP, immunoprecipitation; WCL, whole-cell lysate). (b) Cdc20 interacts with multiple domains on RASSF1A. FLAG-Cdc20 and different fragments of HA-RASSF1A were co-transfected into HEK293 cells. The immunoprecipitate of FLAG-cdc20 was analyzed by immunoblotting. (c) D-box and KEN-box of RASSF1A are dispensable for the interaction with Cdc20. Different D-box deletion mutants of RASSF1A were co-transfected with FLAG-Cdc20 into HEK293 cells and the immunoprecipitation–immunoblotting assay was performed as in (b). (d) Mapping of D-box-dependent and -independent Cdc20-binding domains of RASSF1A. FLAG-Cdc20 and different fragments of HA-RASSF1ADBD+KD were co-transfected into HEK293 cells and the immunoprecipitation–immunoblotting assay was performed as in (b).
RASSF1A inhibits APC/CCdc20 via D-boxes
RASSF1A was suggested to inhibit Cdc20 by sequestering Cdc20 from APC/C previously (Song et al., 2004). It seems to contradict with our present finding on the APC/CCdc20 induction of RASSF1A degradation. Intriguingly, we found that recombinant RASSF1A did inhibit APC/C by in vitro APC/C inhibitory assay (Figure 4a). The finding is in concordance with the findings in previous reports (Song et al., 2004, 2008).

Inhibition of APC/CCdc20 by RASSF1A depends on the D-box clusters. (a) RASSF1A inhibits APC/CCdc20 in vitro. Recombinant MBP-RASSF1A or MBP was added to the in vitro ubiquitination assay in the presence of Cdc20(WT) or Cdc20(7A). The 35S-labeled N-terminal fragment of Cyclin B1 was used as the substrate (left panel). The amount of poly-ubiquitinated Cyclin B1 was quantified and the APC/C activities in each reaction relative to the control reaction containing Cdc20(7A) and MBP were calculated (right panel). (b) Deletion of the D-box clusters abolishes the inhibitory effect of RASSF1A in vitro. In vitro APC/C inhibitory assay was performed with recombinant RASSF1ADBD mutants (left panel) and the activity of APC/C in the reaction was determined (right panel). The loadings of different RASSF1A mutants were determined by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and Coomassie Brilliant Blue (CBB) stain.
We further investigated the mechanism of RASSF1A-mediated inhibition on APC/CCdc20 activity. As RASSF1A contains multiple D-boxes and a KEN-box, we hypothesized that RASSF1A inhibits APC/CCdc20 via D-boxes like APC/C pseudosubstrates Emi1 (Miller et al., 2006). To determine the function of the D-boxes and KEN-box on APC/CCdc20 inhibition, APC/C inhibitory assay was performed using recombinant RASSF1A with different D-boxes being deleted. Upon deletion of any D-box cluster, RASSF1A could not inhibit APC/CCdc20 effectively (Figure 4b). The finding confirmed that the D-boxes are critical for APC/CCdc20 inhibition.
Aurora-dependent phosphorylation of RASSF1A turns RASSF1A from being an inhibitor to a substrate of APC/CCdc20
Our results suggested that RASSF1A is both an inhibitor and a substrate of APC/CCdc20. We then asked how these two distinct functions of RASSF1A are controlled. As this change in RASSF1A function occurs at mitosis, we hypothesized that the inhibitor or substrate property of RASSF1A is controlled by mitotic kinase phosphorylation. Recent studies have reported that RASSF1A is phosphorylated by Aurora A and B (Rong et al., 2007; Song et al., 2009). In this study, we also confirmed their finding that T202 and S203 residues of RASSF1A were the Aurora phosphorylation sites (Supplementary Figure 2). Interestingly, Rong et al. (2007) and Song et al. (2009) showed that phosphorylated form of RASSF1A failed to induce mitotic arrest. Therefore, we postulated that Aurora phosphorylation can change the role of RASSF1A from an inhibitor to a substrate of APC/CCdc20. To address this hypothesis, in vitro ubiquitination reaction was set up using 35S-labeled RASSF1A as substrate (Figure 5a). The in vitro-translated RASSF1A was added to the ubiquitination reaction containing mitotic APC/C (m-APC/C) purified from mitotic HeLa cells in the presence or absence of Aurora A or Aurora B. The result showed that RASSF1A was ubiquitinated by m-APC/C, and strikingly, addition of Aurora A or Aurora B enhanced the ubiquitination of RASSF1A by 7-fold and 14-fold, respectively (Figure 5a, left panel). A control in vitro ubiquitination experiment was set up in the presence of Aurora A or Aurora B with or without the m-APC/C. The result showed that the ubiquitinated form of RASSF1A was absent in the control without m-APC/C (Supplementary Figure 3). The data suggested that the ubiquitination of RASSF1A was APC/C-dependent. To confirm the importance of the phosphorylation on the ubiquitination of RASSF1A, we performed in vitro ubiquitination reaction on wild-type, phospho-null and phospho-mimic mutants of RASSF1A in the presence of Aurora B. The wild-type RASSF1A (WT) and the phospho-mimic RASSF1A (T202E/S203E) were readily ubiquitinated by m-APC/C in the presence of Aurora B. However, the phospho-null RASSF1A (T202A/S203A) was resistant to ubiquitination (Figure 5a, right panel). In addition, rather than RASSF1AWT, phospho-mimic RASSF1A was prone to ubiquitination by m-APC/C without the supplement of Aurora B (Figure 5a, right panel). These data suggested that Aurora kinase phosphorylation of RASSF1A primes APC/C-dependent ubiquitination on RASSF1A. To investigate the role of Aurora-dependent phosphorylation of RASSF1A on its stability in vivo, we performed the in vivo degradation assay of RASSF1A. For this assay, HeLa cells transfected with WT HA-RASSF1A were treated either with or without Aurora inhibitor (ZM447439 or 4-(4′-Benzamidoanilino)-6,7-dimethoxyquinazoline (AI-II)) for 2 h (Mortlock et al., 2005; Heron et al., 2006). Different mutants of HA-RASSF1A were ectopically expressed in mitotic HeLa cells. The protein translation of the cells was blocked by treatment of cycloheximide and cells were collected at different time points for immunoblotting (Figure 5b). The effect of the Aurora inhibitors on Aurora kinases was confirmed by the reduction of phospho-Histone H3 (S-10) level (Crosio et al., 2002). Agreeing with the result of Figure 5a, WT and phospho-mimic (T202E/S203E) RASSF1A was degraded rapidly. However, the phospho-null (T202A/S203A) RASSF1A or RASSF1A in cells pretreated with Aurora inhibitor was relatively stable at the first 60 min after the treatment of cycloheximide. Similar results were obtained from HeLa cells co-transfected with HA-RASSF1A and Cdc20(7A) (Supplementary Figure 4a). Together with the in vitro ubiquitination assay, we clearly demonstrated that Aurora-dependent phosphorylation was essential for APC/CCdc20-mediated RASSF1A ubiquitination and degradation.

Aurora kinases facilitated the ubiquitination and degradation of RASSF1A. (a) Aurora kinases facilitated the ubiquitination of RASSF1A by purified mitotic APC/C in vitro. The in vitro ubiquitination reaction was set up using APC/C purified from mitotic HeLa cells. (Left panel) 35S-labeled RASSF1A was used as substrate. (Right panel) 35S-labeled wild-type or phospho-mutants of RASSF1A were used as substrate. Where indicated, Aurora A or Aurora B was added. (b) Phosphorylation of RASSF1A by Aurora kinases is essential for Cdc20-dependent degradation. HeLa cells were transfected with different mutants of HA-RASSF1A. After 24 h, cells were arrested to mitosis by nocodazole. Where indicated, cells were treated with 2 μM ZM447439 or 2 μM AI-II for 2 h. The cells were then treated with 50 μg/ml cycloheximide (CHX) and collected at indicated time points for immunoblotting. (c) APC/CCdc20 ubiquitinates RASSF1A at DB1 and DB2 in vitro. Wild-type or mutated 35S-labeled RASSF1A was subjected to in vitro ubiquitination assay using mitotic HeLa APC/C and purified Aurora B. RASSF1A was resistant to APC/C ubiquitination when both DB1 and DB2 were deleted. (d) DB1 and DB2 of RASSF1A are essential for Cdc20-dependent degradation in vivo. HeLa cells were transfected with different HA-RASSF1A mutants and treated as in (b). Consistent with in vitro ubiquitination assay, RASSF1ADBD1+2 mutant was resistant to Cdc20-dependent degradation.
As there are six D-boxes on RASSF1A, we further examined which D-box is the degron recognized by APC/CCdc20. The in vitro ubiquitination assay of RASSF1A mutants was carried out in the presence of m-APC/C and Aurora B. In our initial analysis, in vitro-translated 35S-labeled single D-box deletion mutants of RASSF1A (RASSF1ADBD) were used as substrates. Surprisingly, none of the single D-box mutants of RASSF1A showed resistance to m-APC/C-dependent ubiquitination (Figure 5c, left panel). We then generated RASSF1ADBD mutants with different combinations of D-box deletion. The result showed that RASSF1A with DB1 and DB2 deleted was resistant to m-APC/C-dependent ubiquitination (Figure 5c, right panel). The requirement of DB1 and DB2 for Cdc20-dependent degradation of RASSF1A was further confirmed by in vivo degradation assay. Different D-box-deleted mutants of RASSF1A were transfected into HeLa cells and the cells were arrested in mitosis. The degradation of RASSF1A was chased by cycloheximide block (Figure 5d). As expected, HA-RASSF1AWT was degraded rapidly upon cycloheximide treatment. Moreover, HA-RASSF1A mutants without DB1 and DB2 stabilized HA-RASSF1A from Cdc20(7A)-dependent degradation. Similar result was seen in HeLa cells co-transfected with different D-box-deleted mutants of RASSF1A and FLAG-Cdc20(7A) (Supplementary Figure 4b). The data provided clear evidence that DB1 and DB2 of RASSF1A serve as bona fide degrons in the APC/CCdc20-mediated degradation.
In our study, we demonstrated that RASSF1A was ubiquitinated by APC/CCdc20 and the ubiquitination was enhanced by Aurora-dependent phosphorylation. Then, we further determined whether the interaction between RASSF1A and Cdc20 is affected by Aurora phosphorylation. In the report of Song et al. (2009), they proposed that phospho-mimic mutant of RASSF1A did not bind to Cdc20 and this allowed Cdc20 to activate APC/C. However, our result indicated that RASSF1AWT, RASSF1AT202A/S203A and RASSF1AT203E/S203E could immunoprecipitate with ectopically expressed Cdc20 (Supplementary Figure 5a). In addition, we demonstrated that both phospho-mimic and phospho-null mutants of RASSF1A bind to endogenous APC/CCdc20. Cdc20 or Cdc27 were immunoprecipitated from the mitotic HeLa cells transfected with phospho-mutants of RASSF1A. Our result indicated that both endogenous Cdc20 and Cdc27 presented in the RASSF1A immunoprecipitates (Supplementary Figure 5b). Furthermore, we determined the effect of Aurora activity on the interaction between endogenous RASSF1A and Cdc20. Mitotic HeLa cells were treated with MG132, Aurora inhibitor ZM447439 (ZM) or AI-II and the interaction between endogenous RASSF1A and Cdc20 was determined by immunoprecipitation (Supplementary Figure 5c). The result showed that RASSF1A could bind to Cdc20 regardless of the Aurora activity. Then, we asked if the Aurora phosphorylation is responsible for the binding between RASSF1A and Cdc20 in a D-box-independent manner. To address this question, the phospho-mutants of FL-RASSF1ADBD+KD, RASSF1ADBD+KD (1–264) and RASSF1ADBD+KD (141–340) were generated by mutagenesis. Different RASSF1A mutants and Cdc20 were co-transfected into HeLa cells and the binding between RASSF1A mutants and Cdc20 was determined by immunoprecipitation (Supplementary Figure 5d). The result illustrated that Cdc20 co-precipitated with different mutants of RASSF1A irrespective to the phosphorylation site mutation.
RASSF1A regulates APC/C activity and Cyclin A degradation during mitosis
We showed that RASSF1A is an inhibitor of APC/C and it becomes a substrate of APC/CCdc20 after phosphorylating by Aurora kinase. The physiological significance of this regulatory circuit was then investigated. It was shown that APC/C ubiquitinated its substrates sequentially during mitosis (Peters, 2006). As RASSF1A was degraded irrespective of spindle checkpoint, we hypothesized that RASSF1A controls the degradation of APC/C substrates before the onset of spindle checkpoint. Similar to RASSF1A, Cyclin A and Nek2A are degraded by APC/C irrespective of the spindle checkpoint (Geley et al., 2001; Hames et al., 2001; Hayes et al., 2006). Therefore, we determined whether RASSF1A controls the degradation of Cyclin A and Nek2A. RASSF1A was either overexpressed or knocked down in HeLa cells. Cells were then synchronized by double thymidine block and were released into nocodazole. Immunoblottings revealed that the degradation of Cyclin A was delayed in cells transfected with RASSF1A or phospho-null RASSF1A compared with control cells (Figure 6a and Supplementary Figure 6). Degradation of Nek2A was also delayed in cells transfected with RASSF1A (Figure 6a). On the other hand, Cyclin B1 degradation was inhibited by the activation of spindle checkpoint. This indicated that RASSF1A regulates the time of Cyclin A and Nek2A degradation during mitosis. However, the degradation of Cyclin B was regulated by spindle checkpoint. HeLa cells transfected with si-RASSF1A showed significantly less Cyclin A accumulation at early mitosis than control siRNA (Figure 6b). Cyclin B1 also accumulated in cells transfected with si-RASSF1A during the mitotic progression, despite the fact that the level of Cyclin B1 protein was initially lowered in RASSF1A knockdown cells. This concurs with our finding that RASSF1A inhibits the activity of APC/C. In addition, siRNA of RASSF1A did not change the time of degradation of Cyclin A and Nek2A. This suggested that there is another level of regulation of APC/C or its substrates in starting the degradation process (Peters, 2006; Pines, 2006; van Zon and Wolthuis, 2010).

(a) RASSF1A delays Cyclin A degradation during mitosis. HeLa cells transfected with HA-RASSF1A were synchronized by thymidine block and released into nocodazole. Cells were collected at indicated time points for immunoblotting (left panel). Cyclin A and Nek2A were not degraded until 10 h post-thymidine block in cells transfected with HA-RASSF1A, whereas it was degraded at 8 h in control cells. (b) RASSF1A promotes accumulation of Cyclin A. HeLa cells transfected with siRNA targeting RASSF1A were synchronized and analyzed as in Figure 2b. The level of Cyclin A was consistently lower in cells transfected with RASSF1A siRNA than those transfected with control siRNA throughout the assay. (c) D-box-deleted RASSF1A mutants fail to delay the degradation of Cyclin A during mitosis. HeLa cells transfected with D-box-deleted mutants of RASSF1A were synchronized and analyzed as in (b). The immunoblotting was performed (upper panel). Although RASSF1AWT delayed the degradation of Cyclin A after the thymidine block, none of the RASSF1ADBD mutants delayed the degradation of Cyclin A. *Time point when Cyclin A degradation started.
As we showed that RASSF1A regulates the APC/C via D-boxes, we next investigated the effect of RASSF1ADBD mutants on the time of Cyclin A degradation. In concordance with the result of in vitro inhibitory assay, none of the RASSF1ADBD mutant delayed the degradation of Cyclin A during mitosis (Figure 6c). This result suggested that the D-boxes are important for RASSF1A to function as an APC/C inhibitor.
Discussion
RASSF1A inhibits APC/CCdc20 activity at early mitosis
In this study, our data supported a role for RASSF1A as an inhibitor of APC/CCdc20. In a previous study, Song et al. (2004) suggested that RASSF1A could induce G2/M arrest through inhibiting the APC/C activity by sequestering Cdc20 after the dissociation of APC/CCdc20 from Emi1. Here, we further demonstrated that multiple D-boxes in RASSF1A play an important role in inhibiting APC/CCdc20 activity. Using in vitro APC/C inhibitory assay, we showed that RASSF1AWT was more potent in inhibiting the APC/CCdc20 activity, whereas D-box-deleted mutants of RASSF1A were less effective. Also, although degradation of Cyclin A by APC/CCdc20 could be found in the presence of RASSF1AWT, D-box-deleted mutants did not exert an effect on the degradation of Cyclin A in synchronized HeLa cells during mitosis. Particularly, our result suggested that the first two D-boxes (DB1 and DB2) of RASSF1A exert critical effects on its inhibitory function. Among the RASSF1 isoforms, only RASSF1A was shown to be a key tumor suppressor involving in mitosis regulation (Rong et al., 2004; Song et al., 2004). Different from other isoforms such as RASSF1C at the N-terminal, the first two D-boxes of RASSF1A are specific to RASSF1A and are not found in other isoforms. Our new finding provides an explanation in that APC/CCdc20 is preferentially inhibited by RASSF1A. Intriguingly, deletion of the D-boxes and KEN-box did not affect the binding between RASSF1A and Cdc20. However, RASSF1ADBD1−6 failed to inhibit APC/CCdc20. This raised the possibility that RASSF1A does not inhibit Cdc20 by sequestration. Indeed, our data supported the notion that RASSF1A might inhibit APC/CCdc20 as a pseudosubstrate, similar to Mad3p, Acm1 and Emi1 (Miller et al., 2006; Burton and Solomon, 2007; Enquist-Newman et al., 2008). In the model of pseudosubstrate inhibition of APC/C, the inhibitors present a D-box-like structure but, for unknown mechanism, these pseudosubstrates are not ubiquitinated by APC/C (Miller et al., 2006; Burton and Solomon, 2007; Enquist-Newman et al., 2008). Similarly, RASSF1A is not ubiquitinated effectively by APC/C in vitro in the absence of Aurora kinase. Thus, we believe that RASSF1A maintains as a stable pseudosubstrate of APC/C at early mitosis.
Although an earlier report from Liu et al. (2007) did not suggest an interaction between RASSF1A and Cdc20, more recent investigations have highlighted interaction between RASSF1A and Cdc20 (Wang et al., 2008; Song et al., 2009). In this study, we found that RASSF1A can bind to Cdc20 in a D-box-dependent and D-box-independent manner. Specifically, the first two D-boxes at the N-terminus of RASSF1A was shown to be Cdc20-binding domains. In addition, a D-box-independent Cdc20-binding domain was mapped to residues 141–264 of RASSF1A. Our study has revealed an interesting binding strategy between RASSF1A and Cdc20
Phosphorylation by Aurora kinases turns RASSF1A from an inhibitor to a specific substrate of APC/CCdc20
In this study, we demonstrated that Aurora kinases-dependent phosphorylation triggers ubiquitination of RASSF1A by APC/CCdc20. Whereas Aurora A is a well-characterized oncoprotein that promotes mitotic progression, RASSF1A is a tumor suppressor that regulates mitosis. The interplay between an oncoprotein and tumor suppressor in mitosis can be shown in this autoregulatory model. At the beginning of mitosis, RASSF1A inhibits APC/CCdc20. Meanwhile, the accumulation and activation of Aurora A and Aurora B, during the course of mitosis progression, allow phosphorylation of RASSF1A that turn it into a substrate of APC/CCdc20 (Figure 7). Additionally, RASSF1A was shown to activate Aurora A (Liu et al., 2008). Thus, RASSF1A may play a role in allowing the accumulation and activation of Aurora A during mitosis, and this in turn primes RASSF1A to degradation.

Model of RASSF1A regulation during mitosis. At the early mitosis, APC/CCdc20 is sequentially inhibited by Emi1 and RASSF1A. RASSF1A is subsequently phosphorylated by Aurora A and Aurora B accumulating during mitosis. This phosphorylation turns RASSF1A from an inhibitor to a substrate of APC/CCdc20. As a result, RASSF1A is ubiquitinated and targeted for proteasomal degradation. Consequently, the partially activated APC/CCdc20 can ubiquitinate Cyclin A. The whole process occurs before passing the spindle checkpoint.
In this study, we presented data to support that RASSF1A was ubiquitinated by APC/CCdc20 in mitosis. Recent studies suggested that RASSF1A is ubiquitinated by different E3 ligases. The study by Song et al. (2008) showed that RASSF1A was degraded at S phase in U2OS cell line, and the degradation of RASSF1A in S phase was mediated by SCFSkp2 complex. On the other hand, Jiang et al. (2011) pinpointed that RASSF1A was targeted by CUL4A-DDB1 complex during mitosis. Taking together, these studies suggested that RASSF1A is differentially regulated by ubiquitination in the cell cycle.
It was proposed that Aurora A regulates RASSF1A in mitosis by different mechanisms (Rong et al., 2007; Song et al., 2009). In our model, we proposed that Aurora A mediated Cdc20-dependent ubiquitination of RASSF1A in mitosis. This released the RASSF1A-dependent mitotic blockage. In addition, Song et al. (2009) demonstrated that the expression of phospho-mimetic RASSF1A in HeLa cell could overcome the mitotic arrest caused by knocking down of Aurora A. This implied that RASSF1A is a key substrate of Aurora A in regulating the mitotic progression.
To our understanding, this is the first report showing that Aurora kinases enhanced the ubiquitination of APC/C substrate. Our result raises an important argument on whether phosphorylation controls the ubiquitination of APC/C substrates. Previous reports have shown that phosphorylation prevents the ubiquitination of several APC/C substrates (Woodbury and Morgan, 2007; Enquist-Newman et al., 2008). These studies highlighted that phosphorylation may change the conformation of substrates, which in turn hide the D-box from Cdc20 or Cdh1. In the case of RASSF1A, Aurora kinase-dependent phosphorylation may alter its conformation, which allows APC/CCdc20 to ubiquitinate RASSF1A.
In this study, DB1 and DB2 were demonstrated to be the Cdc20-specific degrons on RASSF1A. As discussed before, these two D-boxes are critical for the APC/C inhibitory function of RASSF1A. Thus, DB1 and DB2 are the functional and autoregulatory domains on RASSF1A; they are necessary for RASSF1A to be an APC/C inhibitor, and they are also essential for the degradation of RASS1A. Importantly, as the DB1 and DB2 are specific for RASSF1A, the expression of RASSF1A, not other RASSF1 isoforms, can be specifically regulated during mitosis.
RASSF1A is a specific substrate of Cdc20 but not Cdh1. Although several D-boxes are located at different regions on RASSF1A, our data showed that RASSF1A associates only with Cdc20. In addition to RASSF1A, Acm1 and p21 were shown to be specific targets of APC/CCdc20 (Hames et al., 2001; Amador et al., 2007; Enquist-Newman et al., 2008). Study on Acm1 has suggested that Cdc20 and Cdh1 can recognize D-box in different ways (Enquist-Newman et al., 2008). The D-box at the N-terminal of Acm1 is recognized by Cdc20 only. However, the D-boxes at the middle region of Acm1 bind to Cdh1 at high affinity. This implied that Cdc20 and Cdh1 recognize the substrates by different mechanisms. Therefore, further investigation on the Cdc20-RASSF1A- and Cdc20-Acm1-binding mechanisms may reveal important insights on APC/CCdc20-specific interaction.
The activity of APC/CCdc20 is controlled by spindle assembly checkpoint. Our results suggested that degradation of RASSF1A by APC/CCdc20 can occur in the presence of active spindle checkpoint. In fact, only few substrates of APC/CCdc20, such as Cyclin A and Nek2A, are ubiquitinated in the presence of spindle checkpoint. Recently, Izawa and Pines (2011) demonstrated that Cdc20 cooperates with APC8 to recruit substrate to APC/C in the presence of spindle checkpoint. On the other hand, when the spindle checkpoint is satisfied, Cdc20 cooperates with APC8 and APC3 in recruiting substrate to APC/C. This in turn changes the substrate specificity so that APC/CCdc20 ubiquitinates different substrates at different stages of mitosis(Izawa and Pines, 2011). Therefore, it is conceivable that RASSF1A is recruited by Cdc20 and APC8 to APC/C for ubiquitination.
RASSF1A determines the timely degradation of Cyclin A
RASSF1A determines the time of degradation of APC/C substrates in early mitosis. We demonstrated that RASSF1A regulates the accumulation of Cyclin A and NEK2A. By inhibiting the APC/C, RASSF1A determines the time of degradation of Cyclin A and NEK2A. Using Cyclin A as a model, we propose that degradation of RASSF1A primes the partial activation of APC/C regardless of the spindle checkpoint. The partial activity of APC/C is critical in ubiquitination and degradation of APC/C substrates such as Cyclin A. This step is crucial for the initiation of anaphase (Geley et al., 2001; den Elzen and Pines, 2001).
To conclude, we propose a model that supports an autoregulatory mechanism for the APC/CCdc20 proteasomal activity (Figure 7). At the early mitosis, APC/CCdc20 is inhibited by RASSF1A, whereas phosphorylation of RASSF1A by Aurora kinases turns RASSF1A to a substrate of APC/CCdc20. Consequently, APC/CCdc20 ubiquitinates RASSF1A. This results in the partial activation of APC/CCdc20 that can further ubiquitinate Cyclin A and NEK2A in the presence of spindle checkpoint. After fulfilling the spindle checkpoint, APC/CCdc20 further ubiquitinates its substrates including Cyclin B1 and securin. This model provides a new mechanism for the spatial degradation of APC/CCdc20 substrates.
Materials and methods
Cell culture, cell cycle synchronization and drug treatments
HeLa cells and HEK293 cells were obtained from American Type Culture Collection (Manassas, VA, USA) and were maintained in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum. HeLa cells are synchronized by double thymidine block, thymidine–nocodazole block and nocodazole block and release as described (Fang et al., 1998; Petersen et al., 2000). To inhibit the proteasome in vivo, cells were treated with 10 μM MG-132 for 6 h. To inhibit the Aurora activity, cells were treated with 2 μM ZM447439 (Tocris Bioscience, Bristol, UK) or 2 μM AI-II for 2 h. To chase the protein half-life in vivo, cells were incubated with 50 μg/ml cycloheximide for the indicated time points after 24 h post transfection. Unless specified, all the chemicals were purchased from Calbiochem (Gibbstown, NJ, USA).
Plasmids and recombinant proteins
Coding sequence of Cyclin B1 (N-terminal 1–87), Aurora A, Aurora B, Cdc20 and Cdh1 were amplified from HeLa cDNA library by PCR. The open reading frame of RASSF1A was amplified from pcDNA3.1-RASSF1A (a generous gift of R Dammann, University of Heidelberg, Heidelberg, Germany). All of the genes were cloned into pcDNA3.1 with N-terminal (for Cdc20 and Cdh1) or C-terminal (for RASSF1A, Cyclin B1, Aurora A and Aurora B) hemagglutinin (HA) or FLAG tag. Site-directed mutagenesis was performed using Quikchange site-directed mutagenesis (Stratagene, Santa Clara, CA, USA). Expression vector encoding FLAG-Cdc20(7A) mutant was described (Yudkovsky et al., 2000). To generate MBP-RASSF1A, RASSF1A open reading frame was cloned into pMAL-c2 vector (New England Biolabs, Ipswich, MA, USA). MBP and MBP-RASSF1A was expressed in Escherichia coli (BL21) and purified by amylose resin according to standard procedures (New England Biolabs). The primer sequences are available upon request.
Cell cycle analysis by flow cytometry
Synchronized HeLa cells were collected at the indicated time point for propidium iodide stain and FACS analysis (Amador et al., 2007). HeLa cells fixed with 70% ethanol were stained with propidium iodide. All analyses were performed on a FACSCalibur flow cytometer and Cell Quest software (Becton Dickinson, Franklin Lakes, NJ, USA).
siRNA and transfection
Plasmids or siRNAs were transfected into HeLa or HEK293 cells with Lipofectamine 2000 according to the manufacturer's recommendation (Invitrogen, Carlsbad, CA, USA). The siRNA used in this study included Stealth RNAi targeting Cdc20 (5′-AUGCGAAUGUGUCGAUCACUGGUGC-3′) and Skp2 (5′-AAGGGCUGAAAUGUUCAGCCAAUGG-3′) (Invitrogen) and siRNA targeting RASSF1A (5′-dTdTGACCUCUGUGGCGACUUCA-3′) (Qiagen, Hilden, Germany). To determine protein half-life, RASSF1A was co-transfected with Cdc20(7A) in a 1:4 ratio. To transfect and to synchronize cells at G1/S boundary, HeLa cells were transfected for 8 h after the first thymidine block. Cells were then arrested by thymidine again for 18 h and released into fresh medium with 200 ng/ml nocodazole.
Immunoprecipitation and immunoblotting
Immunoprecipitation and immunoblotting was carried out as previously described (Amador et al., 2007). The antibodies used in this study included rabbit anti-HA (Abcam, UK), anti-Cdc20 (Santa Cruz, Santa Cruz, CA, USA) anti-Aurora A and anti-Aurora B (both from Epitomics, Burlingame, CA, USA), anti-Histone H3 and anti-phospho-Histone H3 (Ser-10) (both from Millipore, Billerica, MA, USA); mouse anti-FLAG (Sigma, St Louis, MO, USA), anti-Cdh1 (Sigma), anti-Cdc27 (Sigma), anti-RASSF1A (Abcam), anti-Cyclin B1 (Abcam, Cambridge, UK), anti-Skp2 (Invitrogen) and anti-Cyclin A (Novocastra, Newcastle upon Tyne, UK); and goat anti-actin (Santa Cruz). The band intensity of the immunoblot was quantified by Quantity One software (Bio-Rad, Hercules, CA, USA).
In vitro ubiquitination assay and APC/C inhibitory assay
In vitro ubiquitination assay and APC/C inhibitory assay were performed as previously described (Braunstein et al., 2007; Reddy et al., 2007). Further details are provided in the Supplementary Materials and Methods.
In vitro kinase assay
Aurora A or Aurora B kinase assay was performed according to the manufacturer's protocol (Cell Signaling Technology, Beverly, MA, USA). Further details are provided in the Supplementary Materials and Methods.
References
Amador V, Ge S, Santamaria PG, Guardavaccaro D, Pagano M . (2007). APC/C(Cdc20) controls the ubiquitin-mediated degradation of p21 in prometaphase. Mol Cell 27: 462–473.
Braunstein I, Miniowitz S, Moshe Y, Hershko A . (2007). Inhibitory factors associated with anaphase-promoting complex/cylosome in mitotic checkpoint. Proc Natl Acad Sci USA 104: 4870–4875.
Burton JL, Solomon MJ . (2007). Mad3p, a pseudosubstrate inhibitor of APCCdc20 in the spindle assembly checkpoint. Genes Dev 21: 655–667.
Carmena M, Ruchaud S, Earnshaw WC . (2009). Making the Auroras glow: regulation of Aurora A and B kinase function by interacting proteins. Curr Opin Cell Biol 21: 796–805.
Crosio C, Fimia GM, Loury R, Kimura M, Okano Y, Zhou H et al. (2002). Mitotic phosphorylation of histone H3: spatio-temporal regulation by mammalian Aurora kinases. Mol Cell Biol 22: 874–885.
den Elzen N, Pines J . (2001). Cyclin A is destroyed in prometaphase and can delay chromosome alignment and anaphase. J Cell Biol 153: 121–136.
Enquist-Newman M, Sullivan M, Morgan DO . (2008). Modulation of the mitotic regulatory network by APC-dependent destruction of the Cdh1 inhibitor Acm1. Mol Cell 30: 437–446.
Fang G, Yu H, Kirschner MW . (1998). Direct binding of CDC20 protein family members activates the anaphase-promoting complex in mitosis and G1. Mol Cell 2: 163–171.
Geley S, Kramer E, Gieffers C, Gannon J, Peters JM, Hunt T . (2001). Anaphase-promoting complex/cyclosome-dependent proteolysis of human cyclin A starts at the beginning of mitosis and is not subject to the spindle assembly checkpoint. J Cell Biol 153: 137–148.
Hames RS, Wattam SL, Yamano H, Bacchieri R, Fry AM . (2001). APC/C-mediated destruction of the centrosomal kinase Nek2A occurs in early mitosis and depends upon a cyclin A-type D-box. EMBO J 20: 7117–7127.
Hayes MJ, Kimata Y, Wattam SL, Lindon C, Mao G, Yamano H et al. (2006). Early mitotic degradation of Nek2A depends on Cdc20-independent interaction with the APC/C. Nat Cell Biol 8: 607–614.
Heron NM, Anderson M, Blowers DP, Breed J, Eden JM, Green S et al. (2006). SAR and inhibitor complex structure determination of a novel class of potent and specific Aurora kinase inhibitors. Bioorg Med Chem Lett 16: 1320–1323.
Izawa D, Pines J . (2011). How APC/C-Cdc20 changes its substrate specificity in mitosis. Nat Cell Biol 13: 223–233.
Jiang L, Rong R, Sheikh MS, Huang Y . (2011). Cullin-4A·DNA damage-binding protein 1 E3 ligase complex targets tumor suppressor RASSF1A for degradation during mitosis. J Biol Chem 286: 6971–6978.
King RW, Glotzer M, Kirschner MW . (1996). Mutagenic analysis of the destruction signal of mitotic cyclins and structural characterization of ubiquitinated intermediates. Mol Biol Cell 7: 1343–1357.
Liu L, Baier K, Dammann R, Pfeifer GP . (2007). The tumor suppressor RASSF1A does not interact with Cdc20, an activator of the anaphase-promoting complex. Cell Cycle 6: 1663–1665.
Liu L, Guo C, Dammann R, Tommasi S, Pfeifer GP . (2008). RASSF1A interacts with and activates the mitotic kinase Aurora-A. Oncogene 27: 6175–6186.
Miller JJ, Summers MK, Hansen DV, Nachury MV, Lehman NL, Loktev A et al. (2006). Emi1 stably binds and inhibits the anaphase-promoting complex/cyclosome as a pseudosubstrate inhibitor. Genes Dev 20: 2410–2420.
Mortlock AA, Keen NJ, Jung FH, Heron NM, Foote KM, Wilkinson RW et al. (2005). Progress in the development of selective inhibitors of aurora kinases. Curr Top Med Chem 5: 807–821.
Peters JM . (2006). The anaphase promoting complex/cyclosome: a machine designed to destroy. Nat Rev Mol Cell Biol 7: 644–656.
Petersen BO, Wagener C, Marinoni F, Kramer ER, Melixetian M, Lazzerini Denchi E et al. (2000). Cell cycle- and cell growth-regulated proteolysis of mammalian CDC6 is dependent on APC-CDH1. Genes Dev 14: 2330–2343.
Pfleger CM, Kirschner MW . (2000). The KEN box: an APC recognition signal distinct from the D box targeted by Cdh1. Genes Dev 14: 655–665.
Pines J . (2006). Mitosis: a matter of getting rid of the right protein at the right time. Trends Cell Biol 16: 55–63.
Reddy SK, Rape M, Margansky WA, Kirschner MW . (2007). Ubiquitination by the anaphase-promoting complex drives spindle checkpoint inactivation. Nature 446: 921–925.
Rong R, Jiang LY, Sheikh MS, Huang Y . (2007). Mitotic kinase Aurora-A phosphorylates RASSF1A and modulates RASSF1A-mediated microtubule interaction and M-phase cell cycle regulation. Oncogene 26: 7700–7708.
Rong R, Jin W, Zhang J, Sheikh MS, Huang Y . (2004). Tumor suppressor RASSF1A is a microtubule-binding protein that stabilizes microtubules and induces G2/M arrest. Oncogene 23: 8216–8230.
Song MS, Song SJ, Ayad NG, Chang JS, Lee JH, Hong HK et al. (2004). The tumour suppressor RASSF1A regulates mitosis by inhibiting the APC-Cdc20 complex. Nat Cell Biol 6: 129–137.
Song MS, Song SJ, Kim SJ, Nakayama K, Nakayama KI, Lim DS . (2008). Skp2 regulates the antiproliferative function of the tumor suppressor RASSF1A via ubiquitin-mediated degradation at the G1-S transition. Oncogene 27: 3176–3185.
Song SJ, Kim SJ, Song MS, Lim DS . (2009). Aurora B-mediated phosphorylation of RASSF1A maintains proper cytokinesis by recruiting Syntaxin16 to the midzone and midbody. Cancer Res 69: 8540–8544.
van Zon W, Wolthuis RM . (2010). Cyclin A and Nek2A: APC/C-Cdc20 substrates invisible to the mitotic spindle checkpoint. Biochem Soc Trans 38: 72–77.
Wang X, Di K, Zhang X, Han HY, Wong YC, Leung SC et al. (2008). Id-1 promotes chromosomal instability through modification of APC/C activity during mitosis in response to microtubule disruption. Oncogene 27: 4456–4466.
Wolthuis R, Clay-Farrace L, van Zon W, Yekezare M, Koop L, Ogink J et al. (2008). Cdc20 and Cks direct the spindle checkpoint-independent destruction of cyclin A. Mol Cell 30: 290–302.
Woodbury EL, Morgan DO . (2007). Cdk and APC activities limit the spindle-stabilizing function of Fin1 to anaphase. Nat Cell Biol 9: 106–112.
Yamano H, Gannon J, Mahbubani H, Hunt T . (2004). Cell cycle-regulated recognition of the destruction box of cyclin B by the APC/C in Xenopus egg extracts. Mol Cell 13: 137–147.
Yu H . (2007). Cdc20: a WD40 activator for a cell cycle degradation machine. Mol Cell 27: 3–16.
Yudkovsky Y, Shteinberg M, Listovsky T, Brandeis M, Hershko A . (2000). Phosphorylation of Cdc20/fizzy negatively regulates the mammalian cyclosome/APC in the mitotic checkpoint. Biochem Biophys Res Commun 271: 299–304.
Zur A, Brandeis M . (2001). Securin degradation is mediated by fzy and fzr, and is required for complete chromatid separation but not for cytokinesis. EMBO J 20: 792–801.
Acknowledgements
This work was supported by Michael and Betty Kadoorie Cancer Genetics Research Program II (MBKCGRPII), Li Ka Shing Institute of Health Science and Hong Kong Research Grant Council (440606).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on the Oncogene website
Supplementary information
Rights and permissions
This work is licensed under the Creative Commons Attribution-NonCommercial-No Derivative Works 3.0 Unported License. To view a copy of this license, visit http://creativecommons.org/licenses/by-nc-nd/3.0/
About this article
Cite this article
Chow, C., Wong, N., Pagano, M. et al. Regulation of APC/CCdc20 activity by RASSF1A–APC/CCdc20 circuitry. Oncogene 31, 1975–1987 (2012). https://doi.org/10.1038/onc.2011.372
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/onc.2011.372
Keywords
This article is cited by
-
New type of interaction between the SARAH domain of the tumour suppressor RASSF1A and its mitotic kinase Aurora A
Scientific Reports (2019)
-
RASSF1A, puppeteer of cellular homeostasis, fights tumorigenesis, and metastasis—an updated review
Cell Death & Disease (2019)
-
The tumor suppressor RASSF10 is upregulated upon contact inhibition and frequently epigenetically silenced in cancer
Oncogenesis (2012)

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}