
Abstract
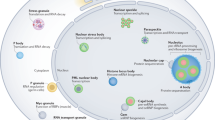
Promyelocytic leukaemia nuclear bodies (PML NBs) are structured protein complexes associated with the nuclear matrix. PML constitutes the scaffold component of NBs and recruits onto these domains a striking variety of proteins, many of which are involved in apoptosis control. Several reports have directly implicated PML in apoptosis and senescence, but the mechanisms by which these are conveyed are still largely unsettled. Recruitment of partner proteins onto NBs is regulated by PML sumolation, a specific post-translational modification also found in many NB-associated proteins. Among these, several are implicated in transcription repression or activation, like the transcriptional repressor Daxx or the transcriptional activator P53. Whether NBs constitute platforms where active sites of enzymatic modifications are carried out, as suggested for P53, sites of intranuclear protein sequestration, as proposed for Daxx or organelles specialized in catabolism, is still debated. A variety of stress-related signalling pathways dramatically modulate the formation of PML NBs, which may provide a clue as to their physiological function.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout


Similar content being viewed by others


References
Anton LC, Schubert U, Bacik I, Princiotta MF, Wearsch PA, Gibbs J, Day P, Realini C, Rechsteiner M, Bennink J and Yewdell J . (1999). J. Cell Biol., 146, 113–124.
Bischof O, Kirsh O, Pearson M, Itahana K, Pelicci PG and Dejean A . (2002). EMBO J., 21, 3358–3369.
Borden KL . (2002). Mol. Cell. Biol., 22, 5259–5269.
D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, Gostissa M, Coen S, Marchetti A, Del Sal G, Piaggio G, Fanciulli M, Appella E and Soddu S . (2002). Nat. Cell Biol., 4, 11–19.
de Thé H, Chomienne C, Lanotte M, Degos L and Dejean A . (1990). Nature, 347, 558–561.
de Thé H, Lavau C, Marchio A, Chomienne C, Degos L and Dejean A . (1991). Cell, 66, 675–684.
Ecsedy JA, Michaelson JS and Leder P . (2003). Mol. Cell. Biol., 23, 950–960.
Emelyanov AV, Kovac CR, Sepulveda MA and Birshtein BK . (2002). J. Biol. Chem., 277, 11156–11164.
Engelhardt OG, Boutell C, Orr A, Ullrich E, Haller O and Everett RD . (2003). Exp. Cell Res., 283, 36–50.
Eskiw CH and Bazett-Jones DP . (2002). Biochem. Cell Biol., 80, 301–310.
Everett RD . (2000). J. Virol., 74, 9994–10005.
Everett RD, Lomonte P, Sternsdorf T, van Driel R and Orr A . (1999). J. Cell Sci., 112, 4581–4588.
Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C and Lowe SW . (2000). Genes Dev., 14, 2015–2027.
Flenghi L, Fagioli M, Tomassoni L, Pileri S, Gambacorta M, Pacini R, Grignani F, Casini T, Ferrucci PF, Martelli MF, Pelicci P-G and Falini B . (1995). Blood, 85, 1871–1880.
Fogal V, Gostissa M, Sandy P, Zacchi P, Sternsdorf T, Jensen K, Pandolfi PP, Will H, Schneider C and Del Sal G . (2000). EMBO J., 19, 6185–6195.
Guiochon-Mantel A, Savouret JF, Quignon F, Delabre K, Milgrom E and de Thé H . (1995). Mol. Endocrinol., 9, 1791–1803.
Guo A, Salomoni P, Luo J, Shih A, Zhong S, Gu W and Paolo Pandolfi P . (2000). Nat. Cell Biol., 2, 730–736.
Henderson BR and Eleftheriou A . (2000). Exp. Cell Res., 256, 213–224.
Hofmann TG and Will H . (2003). Cell Death Differ., 10, 1290–1299.
Hofmann TG, Moller A, Sirma H, Zentgraf H, Taya Y, Droge W, Will H and Schmitz ML . (2002). Nat. Cell Biol., 4, 1–10.
Ishov AM, Sotnikov AG, Negorev D, Vladimirova OV, Neff N, Kamitani T, Yeh E, Strauss J and Maul G . (1999). J. Cell Biol., 147, 221–234.
Jang MS, Ryu SW and Kim E . (2002). Biochem. Biophys. Res. Commun., 295, 495–500.
Jensen K, Shiels C and Freemont PS . (2001). Oncogene, 20, 7223–7233.
Kashuba E, Mattsson K, Klein G and Szekely L . (2003). Mol. Cancer, 2, 18.
Kawai T, Akira S and Reed JC . (2003). Mol. Cell. Biol., 23, 6174–6186.
Kim EJ, Park JS and Um SJ . (2002). J. Biol. Chem., 277, 32020–32028.
Koken MHM, Linares-Cruz G, Quignon F, Viron A, Chelbi-Alix MK, Sobczak-Thépot J, Juhlin L, Degos L, Calvo F and de Thé H . (1995). Oncogene, 10, 1315–1324.
Kurki S, Latonen L and Laiho M . (2003). J. Cell Sci., 116, 3917–3925.
Lafarga M, Berciano MT, Pena E, Mayo I, Castano JG, Bohmann D, Rodrigues JP, Tavanez JP and Carmo-Fonseca M . (2002). Mol. Cell. Biol., 13, 2771–2782.
Lain S, Midgley C, Sparks A, Lane EB and Lane DP . (1999). Exp. Cell Res., 248, 457–472.
Lallemand-Breitenbach V, Zhu J, Puvion F, Koken M, Honore N, Doubeikovsky A, Duprez E, Pandolfi PP, Puvion E, Freemont P and de The H . (2001). J. Exp. Med., 193, 1361–1372.
Langley E, Pearson M, Faretta M, Bauer UM, Frye RA, Minucci S, Pelicci PG and Kouzarides T . (2002). EMBO J., 21, 2383–2396.
Lehembre F, Muller S, Pandolfi PP and Dejean A . (2001). Oncogene, 20, 1–9.
Li H, Leo C, Zhu J, Wu X, O'Neil J, Park EJ and Chen JD . (2000a). Mol. Cell. Biol., 20, 1784–1796.
Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J and Gu W . (2002). Nature, 416, 648–653.
Li R, Pei H, Watson DK and Papas TS . (2000b). Oncogene, 19, 745–753.
Louria-Hayon I, Grossman T, Sionov RV, Alsheich O, Pandolfi PP and Haupt Y . (2003). J. Biol. Chem., 278, 33134–33141.
Mallette F, Goumard S, Gaumont-Leclerc MF, Moiseeva O and Ferbeyre G . (2004). Oncogene, 23, 91–99.
Maul GG, Yu E, Ishov AM and Epstein AL . (1995). J. Cell. Biochem., 59, 498–513.
Megidish T, Xu JH and Xu CW . (2002). J. Biol. Chem., 277, 8255–8259.
Michaelson JS and Leder P . (2003). J. Cell Sci., 116, 345–352.
Michaelson JS, Bader D, Kuo F, Kozak C and Leder P . (1999). Genes Dev., 13, 1918–1923.
Minty A, Dumont X, Kaghad M and Caput D . (2000). J. Biol. Chem., 275, 36316–36323.
Moller A, Sirma H, Hofmann TG, Rueffer S, Klimczak E, Droge W, Will H and Schmitz ML . (2003). Cancer Res., 63, 4310–4314.
Mu ZM, Chin KV, Liu JH, Lozano G and Chang KS . (1994). Mol. Cell. Biol., 14, 6858–6867.
Muller S, Matunis MJ and Dejean A . (1998). EMBO J., 17, 61–70.
Negorev D and Maul GG . (2001). Oncogene, 20, 7234–7242.
Pearson M and Pelicci PG . (2001). Oncogene, 20, 7250–7256.
Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP and Pelicci PG . (2000). Nature, 406, 207–210.
Pokrovskaja K, Mattsson K, Kashuba E, Klein G and Szekely L . (2001). J. Gen. Virol., 82, 345–358.
Puccetti E, Beissert T, Guller S, Li J, Hoelzer D, Ottmann OG and Ruthardt M . (2003). Oncogene, 22, 6900–6908.
Quignon F, de Bels F, Koken M, Feunteun J, Ameisen J-C and de Thé H . (1998). Nat. Genet., 20, 259–265.
Regad T and Chelbi-Alix MK . (2001). Oncogene, 20, 7274–7286.
Reymond A, Meroni G, Fantozzi A, Merla G, Cairo S, Luzi L, Riganelli D, Zanaria E, Messali S, Cainarca S, Guffanti A, Minucci S, Pelicci PG and Ballabio A . (2001). EMBO J., 20, 2140–2151.
Roth MB . (1995). Curr. Opin. Cell Biol., 7, 325–328.
Salomoni P and Pandolfi PP . (2002). Cell, 108, 165–170.
Schmidt D and Muller S . (2002). Proc. Natl. Acad. Sci. USA, 99, 2872–2877.
Seeler JS and Dejean A . (2001). Oncogene, 20, 7243–7249.
Shimoda K, Kamesaki K, Numata A, Aoki K, Matsuda T, Oritani K, Tamiya S, Kato K, Takase K, Imamura R, Yamamoto T, Miyamoto T, Najafuji K, Gondo H, Najafuji S, Nakayama K and Harada M . (2002). J. Immunol., 169, 4707–4711.
Spector DL . (2001). J. Cell Sci., 114, 2891–2893.
Stadler M, Chelbi-Alix MK, Koken MHM, Venturini L, Lee C, Saïb A, Quignon F, Pelicano L, Guillemin M-C, Schindler C and de Thé H . (1995). Oncogene, 11, 2565–2573.
Sternsdorf T, Jensen K, Reich B and Will H . (1999). J. Biol. Chem., 274, 12555–12566.
Stuurman N, de Graaf A, Floore A, Josso A, Humbel B, de Jong L and van Driel R . (1992). J. Cell Sci., 101, 773–784.
Stuurman N, Floore A, Middelkoop E, van Driel R and de Jong L . (1997). Cell. Mol. Biol. Lett., 2, 137–150.
Terris B, Baldin V, Dubois S, Degott C, Flejou JF, Henin D and Dejean A . (1995). Cancer Res., 55, 1590–1597.
Torii S, Egan DA, Evans RA and Reed JC . (1999). EMBO J., 18, 6037–6049.
Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C, Grosveld F and Pandolfi PP . (1998b). Science, 279, 1547–1551.
Wang Z-G, Ruggero D, Ronchetti S, Zhong S, Gaboli M, Rivi R and Pandolfi PP . (1998a). Nat. Genet., 20, 266–272.
Warrell R, de Thé H, Wang Z and Degos L . (1993). N. Engl. J. Med., 329, 177–189.
Wei X, Yu ZK, Ramalingam A, Grossman SR, Yu JH, Bloch DB and Maki CG . (2003). J. Biol. Chem., 278, 29288–29297.
Xirodimas DP, Chisholm J, Desterro JM, Lane DP and Hay RT . (2002). FEBS Lett., 528, 207–211.
Yang S, Kuo C, Bisi JE and Kim MK . (2002). Nat. Cell Biol., 4, 865–870.
Zhong S, Muller S, Ronchetti S, Freemont PS, Dejean A and Pandolfi PP . (2000a). Blood, 95, 2748–2752.
Zhong S, Salomoni P, Ronchetti S, Guo A, Ruggero D and P PP . (2000b). J. Exp. Med., 191, 631–639.
Zhu H, Wu L and Maki CG . (2003). J. Biol. Chem., 278, 49286–49292.
Zhu J, Chen Z, Lallemand-Breitenbach V and de The H . (2002). Nat. Rev. Cancer, 2, 705–714.
Zhu J, Koken MHM, Quignon F, Chelbi-Alix MK, Degos L, Wang ZY, Chen Z and de The H . (1997). Proc. Natl. Acad. Sci. USA, 94, 3978–3983.
Acknowledgements
We thank ARECA for support in some aspects of this work, the Pôle Franco-Chinois de génomique and PRA. YT is supported by a grant from the French Foreign Affairs (MAE).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
This article is cited by
-
Differentiation therapy revisited
Nature Reviews Cancer (2018)
-
PML isoforms IV and V contribute to adenovirus-mediated oncogenic transformation by functionally inhibiting the tumor-suppressor p53
Oncogene (2016)
-
Tumor suppressive pathways in the control of neurogenesis
Cellular and Molecular Life Sciences (2013)
-
Mammalian Clusterin associated protein 1 is an evolutionarily conserved protein required for ciliogenesis
Cilia (2012)
