
Abstract
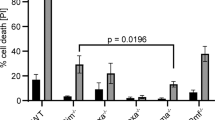
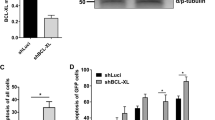
We have examined potential mechanisms by which the Pim-1 kinase acts as a hematopoietic cell survival factor. Enforced expression of the wild type 33 kd (FD/hpim33) and 44 kd (FD/mpim44) Pim-1 proteins in murine factor-dependent FDCP1 cells prolonged survival after withdrawal of IL-3, while expression of a dominant negative Pim-1 protein (FD/pimNT81) shortened survival. Following removal of IL-3 FDCP1 cells exhibited loss of mitochondrial transmembrane potential and production of reactive oxygen species, as determined by flow cytometry analysis. The wild type Pim-1 proteins decreased these changes while the dominant negative protein enhanced mitochondrial dysfunction. The antiapoptotic activity of the kinases could not be attributed to modulation of glutathione, catalase, or superoxide dismutase activities. Both the FD/hpim33 and FD/mpim44 cells maintained expression of bcl-2 mRNA following cytokine removal, while a substantial decrease was seen in FD/neo cells. To modulate Bcl-2 protein levels, a bcl-2 antisense RNA construct was coexpressed with the wild type pim-1 cDNAs. FD/hpim33 cells with low cellular Bcl-2 protein levels had shortened cytokine-independent survival compared with FD/hpim33 clones with high Bcl-2 expression. However survival of FD/mpim44 cells after IL-3 withdrawal was substantially independent of cellular Bcl-2 protein levels. The 33 kd protein delayed, and the 44 kd protein completely prevented enhanced cell death associated with enforced expression of human Bax protein however. Our results suggest that the 33 kd Pim-1 kinase may enhance cell survival through cooperation with and regulation of bcl-2. In addition the 44 kd kinase may regulate the expression or activity of other pro- and anti-apoptotic members of the bcl-2 family.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 50 print issues and online access
$259.00 per year
only $5.18 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout







Similar content being viewed by others

References
Acton, D, Domen J, Jacobs H, Vlaar M, Korsmeyer S and Berns A. . 1992 Curr. Top. Microbiol. Immunol. 182: 293–298.
Ahmed NN, Grimes HL, Bellacosa A, Chan TO and Tsichlis PN. . 1997 Proc. Natl. Acad. Sci. USA 94: 3627–3632.
Allen JD and Berns A. . 1996 Cancer Biol. 7: 299–306.
Amson R, Sigaux F, Przedborski S, Flandrin G, Givol D and Telerman A. . 1989 Proc. Natl. Acad. Sci. USA 86: 8857–8861.
Bossy-Wetzel E, Newmeyer DD and Green DR. . 1998 EMBO J. 17: 37–49.
Breuer M, Wientjens E, Verbeek S, Slebos R and Berns A. . 1991 Cancer Res. 51: 958–963.
Cleveland JL, Troppmair J, Packham G, Askew DS, Lloyd P, Gonzalez-Garcia M, Nunez G, Ihle JN and Rapp UR. . . (1994 Oncogene 9: 2217–2226.
Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y and Greenberg ME. . 1997 Cell 91: 231–241.
del Peso L, Gonzalez-Garcia M, Page C, Herrera R and Nunez G. . 1997 Science 278: 687–689.
Domen J, van der Lugt NMT, Laird PW, Saris CJM, Clarke AR, Hooper ML and Berns A. . 1993 Blood 82: 1445–1452.
Fairbairn LJ, Cowling GJ, Reipert BM and Dexter TM. . 1993 Cell 74: 823–832.
Feldman JD, Vician L, Crispino M, Tocco G, Marcheselli VL, Bazan NG, Baudry M and Herschman HR. . 1998 J. Biol. Chem. 273: 16535–16543.
Gotoh N, Tojo A and Shibuya M. . 1996 EMBO J. 15: 6197–6204.
Hedley D and Chow S. . 1994 Methods Cell Biol. 42: 31–44.
Hirsch T, Marzo I and Kroemer G. . 1997 Biosci. Rep. 17: 67–76.
Hockenbery DM, Oltvai ZN, Yin XM, Milliman CL and Korsmeyer SJ. . 1993 Cell 75: 241–251.
Kinoshita T, Yokota T, Arai K and Miyajima A. . 1995a Oncogene 10: 2207–2212.
Kinoshita T, Yokota T, Arai K and Miyajima A. . 1995b EMBO J. 14: 266–275.
Kinoshita T, Shirouzu M, Kamiya A, Hashimoto K, Yokoyama S and Miyajima A. . 1997 Oncogene 15: 619–627.
Klampfer L, Zhang J, Zelenetz AO, Uchida H and Nimer SD. . 1996 Proc. Natl. Acad. Sci. USA 93: 14059–14064.
Kluck RM, Bossy-Wetzel E, Green DR and Newmeyer DD. . 1997 Science 275: 1132–1136.
Knudson CM and Korsmeyer SJ. . 1997 Nature Genetics 16: 358–363.
Korsmeyer SJ, Shutter JR, Veis DJ, Merry DE and Oltvai ZN. . 1993 Semin. Cancer Biol. 4: 327–332.
Krajewski S, Krajewska M, Shakaik A, Miyashita T, Wang HG and Reed JC. . 1994 Am. J. Pathol. 145: 1323–1336.
Leverson JD, Koskinen PJ, Orrico FC, Rainio EM, Jalkanen KJ, Dash AB, Eisenman RN and Ness SA. . 1998 Mol. Cell. 2: 417–425.
Lilly M, Le T, Holland P and Hendrickson S. . 1992 Oncogene 7: 727–732.
Lilly M and Kraft A. . 1997 Cancer Res. 57: 5348–5355.
Meeker TC, Nagarijan L, ar-Rushdi A, Rovera G, Huebner K and Croce C. . 1987 Oncogene Res. 1: 87–101.
Miller AD and Rosman G. . 1989 Biotechniques 7: 980–990.
Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Liebermann Da, Hoffman B and Reed JC. . 1994 Oncogene 9: 1799–1805.
Northrop JP, Ullman KS and Crabtree GR. . 1993 J. Biol. Chem. 268: 2917–2923.
Nunez G, London L, Hockenberry D, Alexander M, McKearn JP and Korsmeyer SJ. . 1990 J. Immunol. 144: 3602–3610.
Oppenheim RW. . 1997 Adv. Neurol. 72: 69–78.
Ou P and Wolff SP. . 1996 J. Biochem. Biophys. Methods 31: 59–67.
Packham G, Ashmun RA and Cleveland JL. . 1996 J. Immunol. 156: 2792–2800.
Polotskaya A, Zhao Y, Lilly M and Kraft A. . 1993 Cell Growth Differ. 4: 523–531.
Rinaudo MS, Su K, Falk LA, Halder S and Mufson RA. . 1995 Blood 86: 80–88.
Sakai I and Kraft AS. . 1997 J. Biol. Chem. 272: 12350–12358.
Salomoni P, Perrotti D, Martinez R, Franceschi C and Calabretta B. . 1997 Proc. Natl. Acad. Sci. USA 94: 3296–3301.
Sanchez-Garcia I and Grutz G. . 1995 Proc. Natl. Acad. Sci. USA 92: 5287–5291.
Saris CJM, Domen J and Berns A. . 1991 EMBO J. 10: 655–664.
Sato N, Sakamaki K, Terada N, Arai K and Miyajima A. . 1993 EMBO J. 12: 4181–4189.
Shimizu S, Eguchi Y, Kamiike W, Funahashi Y, Mignon A, Lacronique V, Matsada H and Tsujimoto Y. . 1998 Proc. Natl. Acad. Sci. USA 95: 1455–1459.
van den Hoff MJ, Moorman AF and Lamers WH. . 1992 Nucleic. Acids Res. 20: 2902.
van der Lugt NMT, Domen J, Verhoeven E, Linders K, van der Gulden H, Allen J and Berns A. . 1995 EMBO J. 11: 2536–2544.
van Lohuizen M, Verbeek S, Krimpenfort P, Domen J, Saris C, Radaszkiewicz T and Berns A. . 1989 Cell 56: 673–682.
Wang HG, Rapp UR and Reed JC. . 1996 Cell 87: 629–638.
Wingett D, Long A, Kelleher D and Magnuson NS. . 1996 J. Immunol. 156: 549–557.
Yang E, Zha J, Jockel J, Boise LH, Thompson CB and Korsmeyher SJ. . 1995 Cell 80: 285–291.
Zamzami N, Marchetti P, Castedo M, Decaudin D, Macho A, Hirsch T, Susin SA, Petit PX, Mignotte B and Kroemer G. . 1995 J. Exp. Med. 182: 367–377.
Zamzami N, Susin SA, Marchetti P, Hirsch T, Gomez-Monterrey I, Castedo M and Kroemer G. . 1996 J. Exp. Med. 183: 1293–1295.
Acknowledgements
This work was supported in part by NIH grants CA45672 (M Lilly) and DK44741 (A Kraft), as well as by funds from the Academy of Finland (PJ Koskinen).
Author information
Authors and Affiliations
Rights and permissions
About this article
Cite this article
Lilly, M., Sandholm, J., Cooper, J. et al. The PIM-1 serine kinase prolongs survival and inhibits apoptosis-related mitochondrial dysfunction in part through a bcl-2-dependent pathway. Oncogene 18, 4022–4031 (1999). https://doi.org/10.1038/sj.onc.1202741
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.onc.1202741
Keywords
This article is cited by
-
PIM kinases inhibit AMPK activation and promote tumorigenicity by phosphorylating LKB1
Cell Communication and Signaling (2021)
-
The role of G-CSF neuroprotective effects in neonatal hypoxic-ischemic encephalopathy (HIE): current status
Journal of Neuroinflammation (2021)
-
PIM kinases alter mitochondrial dynamics and chemosensitivity in lung cancer
Oncogene (2020)
-
Phosphorylation of NFATC1 at PIM1 target sites is essential for its ability to promote prostate cancer cell migration and invasion
Cell Communication and Signaling (2019)
-
Safety profiling of genetically engineered Pim-1 kinase overexpression for oncogenicity risk in human c-kit+ cardiac interstitial cells
Gene Therapy (2019)
