
Key Points
-
Many recent viral pandemics have been attributed to the ability of some RNA viruses, for example HIV, dengue virus and possibly the severe acute respiratory syndrome (SARS) coronavirus, to change their host range to include humans. The authors discuss the mechanisms of host-range alteration used by a selection of viruses, including Venezuelan equine and Japanese encephalitis viruses (VEEV and JEV, respectively), dengue virus and West Nile virus (WNV).
-
Venezuelan equine encephalitis (VEE) was first recognized as a disease of horses, donkeys and mules in northern South America during the mid 1930s, but there has been renewed interest in this virus because of its potential as a biological weapon. Molecular analysis of epidemic strains — which exploit horses for amplification — and comparison with strains that do not cause epidemic disease, have shown that a few amino-acid mutations can affect host-range alteration. Changes on the surface of the VEE virion seem to be important for these host range changes.
-
JEV causes epidemics of encephalitis in India, Korea, China, South-East Asia and Indonesia. The disease affects children, and is associated with a mortality rate of greater than 20%. However, unlike VEEV, there is no evidence that JEV undergoes mutation and selection to replicate in different hosts. Pigs amplify transmission in peridomestic settings, and migratory birds have a role in dispersion of JEV. Although different genotypes have been isolated, their relevance to pathology and host range is unclear.
-
WNV is now endemic in the United States after first emerging in New York in 1999. WNV has a very broad host range. Forty-nine species of mosquitoes and ticks, and 225 species of birds are susceptible to infection. Other hosts include horses, cattle, llamas, alligators, cats, dogs, wolves and sheep. Transmission of WNV among these species has not been reported. Although humans are probably dead-end hosts, infection with WNV can cause severe disease.
-
Dengue viruses are very important human arboviral pathogens and use humans as reservoir hosts. Aedes aegypti and Aedes albopictus mosquitoes are the most common vectors in urban settings. It is thought that the human epidemic form of dengue virus evolved in the last 2000 years, and genetic analysis indicates that mutations have resulted in adaptation to the urban mosquito host. However, links between mutations and human pathogenicity have not been established.
-
Finally, the authors discuss how host-range changes can be studied experimentally. Cell-culture model systems can be used to find mutations that correlate with virus fitness and adaptation in different host strains. Viruses that replicate in useful laboratory animal models can also be studied in whole animal hosts.
Abstract
Many pandemics have been attributed to the ability of some RNA viruses to change their host range to include humans. Here, we review the mechanisms of disease emergence that are related to the host-range specificity of selected mosquito-borne alphaviruses and flaviviruses. We discuss viruses of medical importance, including Venezuelan equine and Japanese encephalitis viruses, dengue viruses and West Nile viruses.
Similar content being viewed by others

Main
RNA viruses, including HIV1,2, dengue virus (DENV)3,4 and possibly the severe acute respiratory syndrome (SARS) coronavirus5,6,7, have caused recent pandemics by changing their host range to amplify in humans. Mosquito-borne alphaviruses and flaviviruses belong to the arthropod-borne viruses (arboviruses), and both have a positive-sense single-stranded RNA genome. Most arboviral infections are asymptomatic, or present with an influenza-like illness. However, several mosquito-borne alphaviruses and flaviviruses are important human pathogens that cause central nervous system disease, coma or death (Table 1).
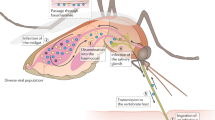
Arboviruses require a host (usually a bird or small mammal) in which they replicate, and a vector, such as a mosquito, for transmission to other organisms. Female mosquitoes ingest virus from the blood of an infected animal. On biting another animal the mosquito transfers the virus through saliva into the new host. Birds are the most common arbovirus hosts, whereas humans and other animals such as horses are usually dead-end hosts — they do not transmit the virus to others in the 'herd', and cannot function as a reservoir for reinfection of mosquitoes. Infection of dead-end hosts can, however, lead to clinical disease (Table 1). In this review, we focus on selected viruses such as Venezuelan equine and Japanese encephalitis viruses (VEEV and JEV, respectively), which cause epidemics by adapting to domestic animals and exploiting them as amplification hosts. We discuss other mosquito-borne viruses including West Nile virus (WNV) and DENV, which cause neurological disease. We consider the patterns and history of host- and geographical-range alterations that lead to disease emergence, and the experimental model systems that are used to study the evolutionary constraints on arbovirus host-range changes.
Venezuelan equine encephalitis virus
Venezuelan equine encephalitis (VEE) was first recognized as a disease of horses, donkeys and mules in South America during the mid-1930s. The VEEV genome encodes four non-structural proteins that participate in genome replication and protein processing, and generates a subgenomic mRNA (26S), which is translated into three main structural proteins. These structural proteins and the positive sense 11.4-kB RNA genome comprise virus particles. Recently, interest in VEEV has been renewed, because it has been developed as an efficient, stable biological weapon that is infectious by aerosol and that is easily produced in large quantities8.
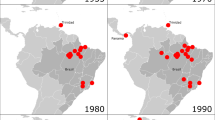
Epidemiology of epizootics. VEEV was isolated in 1938 from a post-mortem brain specimen of a horse with encephalitis9,10, but it was only in the late 1950s that the VEEV was identified as a cause of human disease — presenting as a febrile illness, sometimes accompanied by neurological manifestations and occasional mortality11,12. Until 1995, periodic but unpredictable outbreaks of VEE (some involving hundreds of thousands of equine and human cases) were mostly restricted to Peru, Ecuador, Venezuela and Colombia (Fig. 1). (Human cases were only recognized retrospectively for the earliest outbreaks.) Widespread and long-lasting VEE outbreaks did occur — one EPIZOOTIC began in El Salvador and Guatemala in 1969 and spread through most of Central America and Mexico, reaching Texas in the United States in 1971. However, after 1973, VEE outbreaks and epizootic STRAINS seemed to disappear for 19 years. There were many reasons for this long period of viral inactivity. Equine encephalitis has a high mortality rate, and those animals that survive usually produce protective neutralizing antibodies and are immune to reinfection13,14. Insufficient populations of mosquito vectors also limited incidence of the disease. However, in 1992, an outbreak of VEE occurred in Venezuela (1992—1993 Venezuelan epizootic/epidemic)15, and in 1995, there were epizootics/epidemics in Venezuela and Colombia16, as well as small equine epizootics in southern Mexico in 1993 and 1996 (Ref. 17).

Map showing the locations of all major Venezuelan equine encephalitis (VEE) outbreaks in the Americas (regions shaded purple and labelled with text). The date of the outbreak (year) and the VEE virus (VEEV) subtypes that caused the outbreak are shown. Symbols represent locations from which enzootic VEEV-complex viruses have been isolated, with enzootic subtypes indicated in parentheses.
After identification of VEEV as a cause of human disease, experimental animal models revealed that equine infection results in a high titre VIRAEMIA; the animals therefore serve as highly efficient amplification hosts in the presence of abundant competent mosquito vectors12 (Box 1). In agricultural settings, this efficient amplification facilitates human infection. At the same time, viruses with similar antigens, including the VEE serocomplex of alphaviruses (Fig. 1), were discovered in permanent SYLVATIC cycles in several tropical and subtropical regions of South and Central America, as well as in Mexico and Florida. These ENZOOTIC strains differed from the strains that were isolated during equine epizootics and epidemics; they were antigenically distinct and generated little or no viraemia in experimentally infected horses. It was therefore thought that enzootic strains were incapable of causing epidemics, although fatal infections documented in Panama and Mexico established their virulence as human pathogens18,19. In the late 1960s, antigenic studies shed light on the relationship between epizootic and enzootic VEEV strains. The strains isolated during major epizootics were antigenically similar, and were classified into SUBTYPES IAB and IC. The enzootic strains were antigenically distinct from the epizootic strains and were more diverse. The enzootic strains were grouped into subtypes ID—IF and II—VI, and were later classified as several different species12,20 (Table 2). As it was initially believed that enzootic VEEV strains were unrelated to VEE outbreaks, because they were incapable of amplification in equines and were never isolated during major epidemics, the source of the subtype IAB and IC strains that initiated VEE epizootics and epidemics remained unknown19.
Advances in viral molecular genetics, phylogenetic methodology and computational techniques have provided some answers. Initially, RNA oligonucleotide fingerprinting showed that some enzootic strains in subtype ID were more closely related to certain subtype IC epizootic strains than was indicated by their antigenic properties21. Later, genome sequencing confirmed this similarity22. Phylogenetic studies concluded that the enzootic subtype ID strains were ancestors of the epizootic viruses — now known to have evolved independently on at least three occasions (Fig. 2). This led to the prediction, in 1992, that additional epizootics would follow despite a 19-year hiatus23. In the case of the 1992—1993 Venezuelan outbreak, and both Mexican outbreaks, phylogenetic studies identified SYMPATRIC, closely related enzootic strains that seemed to represent the epizootic progenitors. The 1995 Venezuelan/Colombian isolates were almost identical to strains isolated in the same regions during a 1962—1964 epizootic/epidemic, suggesting either release from a laboratory source or unprecedented genetic stability in a cryptic, natural transmission cycle24.

Phylogenetic tree showing the evolutionary history of Venezuelan equine encephalitis virus (VEEV) emergence derived from gene sequences encoding the PE2 envelope glycoprotein precursor (1,677 nucleotides) using neighbour-joining analysis. VEEV strains are denoted by subtype, followed by country and strain designation. Epizootic strains isolated during equine epizootics are shown in red (VEEV subtypes IAB, IC and IE). The six main enzootic lineages of VEEV are labelled in black. The ancestral phenotypes (enzootic or epizootic) were reconstructed by minimizing phenotypic changes in the branches (treating the phenotype as a character with the most parsimonious reconstruction). Numbers indicate bootstrap values — a measure of how probable the groupings represent descendants of a common ancestor. Reproduced with permission from Ref. 27© (2002) American Society for Microbiology.
Molecular determinants of emergence. Enzootic and epizootic strains of VEEV use different vertebrate hosts and mosquito vectors (Box 1). Therefore, understanding mechanisms of VEE emergence by antigenic shifts and the acquisition of the equine viraemia phenotype requires knowledge of the host-range changes and their genetic basis. Recently, the molecular changes that led to the evolution of epizootic strains from enzootic progenitors have begun to be determined. Analysis of related enzootic ID and epizootic IC strains from western Venezuela facilitated the identification of putative genetic determinants of the epizootic phenotype25. Also, amino-acid sequences of the envelope glycoprotein revealed a common pattern of positive charge (arginine (Arg) or lysine (Lys)) substitutions in a region that was probably located on the surface of the viral particle, which might encode the antigenic determinants26,27 (Box 2). Preliminary studies in which these mutations were introduced into the envelope protein of an enzootic ID strain indicate that an Arg or Lys residue at amino-acid position 213 is the main determinant of the equine viraemia phenotype. Interestingly, these mutations also change the serotype from ID to IAB/IC, explaining the correlation between VEEV serotype and epizootic potential. This single amino-acid substitution, which has a major effect on the vertebrate host range, has resulted in the repeated occurrence of VEE emergence over the past 75 years. This period correlates with the introduction of equines into the New World from Europe, and the establishment of equine populations that are large enough to mediate efficient amplification. RNA viruses, including alphaviruses, have very high mutation frequencies because their error-prone RNA-dependent RNA polymerases lack proof-reading ability28. Estimates of mutation frequency for a closely related alphavirus, eastern equine encephalitis virus (EEEV), are approximately 10−4 substitutions per nucleotide. This indicates that the single mutation leading to the Lys or Arg substitution at position 213 in the envelope glycoprotein E2 occurs regularly in nature, because VEEV populations in mosquitoes and vertebrate reservoir hosts usually exceed 104 plaque-forming units (pfu) ml−1.
An interesting exception to the association between equine VEEV amplification and epizootic transmission is the recent mechanism of equine disease emergence in Mexico17. Although phylogenetic studies support the recent acquisition of the equine virulence phenotype by local, enzootic subtype IE strains27,29, the link between neuroinvasion and viraemia — which occurs in all other epizootic strains (subtypes IAB and IC) — is broken in the Mexican strains isolated from encephalitic horses in 1993 and 1996. These VEEV strains cause equine encephalitis in the absence of high-titre viraemia, which indicates increased neurotropism, as high plasma levels of circulating VEEV are usually necessary for cerebral invasion30. The lack of equine amplification by these strains probably explains their failure to spread to the United States and Central America, despite similar ecological conditions and a geographical position in the path of the widespread 1969—1971 subtype IAB outbreak. Other epizootic amplification hosts are being sought in Mexico to explain the sudden emergence of VEE in 1993, but selection by deforestation for adaption to an alternative, non-sylvatic vector may also be involved31.
Mosquito host range and emergence. As well as adaptation to equine amplification, VEEV emergence from enzootic progenitors requires an alteration in the mosquito-vector host range. All seven of the vector species identified in enzootic VEEV cycles are members of the Culex (Melanoconion) subgenus of mosquitoes, and all of these mosquitoes are also members of the Spissipes section (comprising 23 species) of this subgenus32. The ecological and physiological properties that facilitate efficient transmission of enzootic VEEV by these mosquitoes are unknown, but might include their relative population stability, as they inhabit areas around permanent water sources. However, during outbreaks, floodwater mosquitoes of genera such as Ochlerotatus (formerly named Aedes) and Psorophora — which show large seasonal changes in population density — are the main transmission vectors of epizootic VEEV. Outbreaks often occur in desert-like habitats — such as the Guajira Peninsula of Venezuela and Colombia — at times of unusually heavy rain. Similar climatic events might contribute to the emergence of other arboviruses such as Rift Valley fever virus, which, unlike VEEV, is efficiently maintained by transovarial transmission33. Studies showing that Ochlerotatus taeniorhynchus (probably the most important epizootic vector in coastal outbreaks) has a higher oral susceptibility to epizootic (subtype IAB) compared with enzootic (subtype IE) VEEV indicate that viral adaptation to epizootic mosquito vectors might also be important for VEE emergence31,34. Recently, these findings were extended to the subtype ID enzootic viruses (putative epizootic progenitors), supporting the role of adaptation to mosquito vectors in VEE emergence31,35. Interestingly, the same genetic determinant, the E2-spike envelope glycoprotein, seems to regulate adaptation to both mosquito and equine hosts35. Studies are now underway to determine if the same amino-acid substitutions mediate both host-range changes.
VEEV emergence — unanswered questions. There are many important unanswered questions about VEEV emergence. Why has only one of the closely related enzootic VEEV lineages (Fig. 2) apparently generated all epizootic IAB and IC strains responsible for major outbreaks? Do slight differences in the gene and protein profiles of other enzootic VEEV lineages prevent adaptation to equine hosts and/or epizootic vectors by altering the effect of minor genetic mutations? As equine populations decline in Latin America, and human and domestic animal populations increase, will the scale and frequency of VEE epizootics decrease? Can humans or domestic animals such as cattle replace equines as amplification hosts? Humans can sustain levels of viraemia similar to those of equines, but are probably exposed to mosquito vectors less frequently than domestic animals16,36. Cattle, dogs and other domestic animals are susceptible to infection with VEEV, but levels of viraemias are low, and adaptation would probably be required for amplification in these hosts. During 1995, epidemic VEE occurred in regions of Venezuela lacking equine populations, but with abundant goats, sheep and people, implicating humans or other animals as amplification hosts (G. Medina and N. Perez, personal communication). Can epizootic VEEV strains persist in nature in the absence of efficient equine amplification and transmission? Is epizootic adaptation to equine and mosquito hosts species-specific, resulting in a loss of fitness for the sylvatic hosts that are required for permanent viral circulation? Further research is needed to predict the future impact of VEE, and to design effective measures to prevent and control natural emergence events and possible terrorist introductions.
There are other interesting questions about EEEV and western equine encephalitis virus (WEEV). Unlike VEEV, these viruses do not amplify efficiently in equines (they produce low levels of viraemia), and epizootics involve avian hosts and usually occur only in close proximity to their enzootic swamp (EEEV)37 or agricultural (WEEV)38 habitats. The adaptation of these viruses to equine hosts would have profound public and veterinary health implications. The recombinant ancestors of WEEV are EEEV- and Sindbis-like alphaviruses, both of which use avian enzootic hosts. However, EEEV, but not Sindbis, causes equine and human encephalitis, indicating that the non-structural and/or capsid-protein genes or cis-acting RNA sequences are important determinants of this pathogenic phenotype39.
Japanese encephalitis virus
Although JEV is an arbovirus, it is a member of the genus Flavivirus, family Flaviviridae. The genome of JEV is similar to that of VEEV, as it comprises a single-stranded, positive-sense RNA molecule, approximately 11,000 nucleotides in length. However, in contrast with alphaviruses, the flavivirus genome has no 3′poly(A) tail. Structural proteins, including a single envelope protein, are encoded by the 5′ quarter of the genome.
Japanese encephalitis — the disease. Although Japanese encephalitis (JE) has been recognized as a disease since the 1870s (Ref. 40), the JEV was first isolated in 1935 in Tokyo, Japan, from the brain of a fatal human case of encephalitis41. JEV is the largest worldwide cause of epidemic encephalitis. The virus causes epidemics of paediatric encephalitis, mainly in India, Korea, China, south-east Asia and Indonesia. Owing to its large geographical range, more than two billion people are at risk of infection, and case-fatality rates often exceed 20%. Approximately 50,000 cases occur each year, of which 15,000 are fatal; importantly, up to 50% of those who survive the disease suffer from neurological sequelae that last from months to years. The ratio of apparent to inapparent (asymptomatic) infection varies from 1 in 50 to 1 in 400, depending on the geographical area. For example, in northern Thailand, cases of encephalitis caused by JEV have been diagnosed each year since the late 1960s, with most cases occurring during the rainy season in June, July and August. The fatality rate of virologically confirmed cases is 33%, and, in Thailand, about 1 in 300 humans that are infected with JEV develop encephalitis.
Similar to VEEV, JEV is also a veterinary problem and horses can succumb to encephalitis. However, unlike VEEV, equines are dead-end hosts for JEV, as the level of viraemia is too low to infect mosquitoes. Similarly, an unusually wide range of animals, including birds, dogs, bats and snakes are dead-end hosts that are unable to infect mosquitoes. Pigs and birds are the major amplifying hosts of JEV, although infection usually does not produce clinical disease. These animals do, however, develop high-titre viraemias, which provide an excellent source of infection for mosquitoes. In parts of Asia where pigs are often kept adjacent to human dwellings, these animals are an important source of viral amplification and significantly enhance human exposure and infection. Also, as most domestic pigs are slaughtered by 16 months of age, annual birth cohorts provide a population of animals that are susceptible to infection42. As JEV does occur in areas of Asia where pig populations are low, they are not essential hosts for the enzootic transmission cycle. Interestingly, in central Java, where there are few pigs, it seems that cattle, which are normally considered a dead-end host, might be involved in the natural transmission cycle of JEV43. Migratory birds allow JEV to travel large distances. Unlike VEEV, there is no evidence that enzootic JEV requires adaptation (mutation and selection for replication in pigs or birds) to initiate amplification in these epizootic amplification hosts.
The principal vectors of JEV are Culex tritaeniorhynchus mosquitoes, and maximum virus isolation rates from mosquitoes occur during late July, concurrent with human and equine epidemics. In Malaysia, both Culex gelidus and C. tritaeniorhynchus are important vectors. Other mosquito vectors include members of the Aedes, Anopheles, Mansonia and Armigeres species.
Antigenicity and genotype of JEV. Studies have indicated that there are antigenic and genetic differences among JEV strains, with at least four genotypes and five antigenic subtypes44,45,46. These genotypes and subtypes might correspond, although this has not been experimentally proven. The differences in JEV genotypes might have arisen in response to geographical variation in mosquito vectors or amplifying hosts. The relevance to human disease of the genotypic and antigenic differences among JEV strains is unknown. Early genetic studies used RNA oligonucleotide fingerprinting to identify genetic differences; however, determination of the nucleotide sequence of the first JEV genome in 1987 (strain JaOArs892, genotype III)47 facilitated comprehensive genetic studies. Representative genomic sequences of the four genotypes have been determined, and results are consistent irrespective of the technique used (T1 mapping, partial or complete genome sequencing). The genotypes show different levels of geographical clustering (Fig. 3). Genotype I is found in Australia, Japan, Korea, northern Thailand and Cambodia; genotype II is found in Australia, southern Thailand, Malaysia, Sarawak and Indonesia; genotype III is found throughout Asia and genotype IV is found only in Indonesia. Initial studies suggested that there were genetic differences between strains associated with endemic (round the year) disease in tropical regions of Asia such as Malaysia, Indonesia and the Philippines, compared with epidemic (summer-only) disease in temperate regions of Asia (for example, Japan, Taiwan, Korea and China). However, this has not been confirmed, and differences in endemic and epidemic disease are probably due to climatic variation. JE is an emerging disease, and its geographical distribution is increasing. Australia is the latest country in which JEV has been isolated, and the virus was probably introduced by viraemic migratory birds or wind-borne infected mosquitoes.

a | Neighbour-joining phylogeny of complete Japanese encephalitis virus (JEV) genomes, with a representative strain from other viruses in the JEV serogroup outgrouped using Dengue-2 strain New Guinea C (Den2 NGC). Indonesian isolates are shown in red. b | Neighbour-joining phylogeny of envelope genes, outgrouped using Murray Valley encephalitis (MVE) strain 1-51. Indonesian isolates are shown in red. The tree was constructed using more than 200 isolates from all geographical areas, but for clarity, only a representative isolate of each genotype from each geographical area is shown. Genotypes (I—V) are shown on the right of each tree. Bootstrap values, given as a percentage of 1,000 replicates, are indicated. KUN, Kunjin; SLE, St Louis encephalitis; WN, West Nile. Reproduced with permission from Ref. 49© (2003) American Society for Microbiology.
Phylogeny of JEV. Most phylogenetic studies of the genus Flavivirus imply that the flaviviruses originated in Africa and then spread to other continents48. Phylogenetic studies of JEV reveal that genotype IV is most divergent, with up to 20% nucleotide and 6.5% amino-acid variation compared with genotype I, whereas the other three genotypes differ by no more than 12% at the nucleotide and 3.5% at the amino-acid levels49. The phylogeny of JEV (Fig. 3) indicates that genotype IV, which is found only in Indonesia, is ancestral. Genotypes II and III have also been found, sometimes concurrently, in the Indonesia/Malaysia region. Introduction of JEV into Badu Island in the Torres Straits, Australia, in 1995 involved genotype II, whereas the subsequent introduction of JEV into northern Australia in 1998 involved genotype I. Genotype III is most commonly isolated, and is found throughout Asia (but not Australia). Similarly, genotype III was the only genotype found in Japan until 1998, when genotype I was first isolated. Overall, these results indicate that JEV originated in the Indonesia/Malaysia region and subsequently spread to surrounding areas and countries.
Emergence of JEV. The rate of evolution of JEV indicates that it is a relatively 'young' virus, with genotype IV diverging from the progenitor ancestral virus approximately 350 years ago, with more recent divergence of other genotypes49. It is thought that migratory birds might be important in the emergence of JEV. The Asiatic cattle egret (Bubulcus ibis coromandus) is an important amplifying host, and its geographical range increased in Asia during the nineteenth century because of changes in agricultural practices50. This coincided with the evolution and spread of recent JEV genotypes. It is also tempting to hypothesize that the Second World War had an important role in JEV emergence, because isolation of JEV strains had been restricted to Japan prior to this conflict. Some important questions remain. Why is genotype IV restricted to the Indonesian/Malaysian region, whereas recent genotypes are geographically more widespread? Genotype IV has only been identified once (in 1981), from five mosquito isolates, and its ability to cause human disease is unproven. Although it is thought that genotype IV existed prior to 1981, this has not been confirmed. The sequence of the structural protein genes of a 1952 strain (Muar) from Malaysia51 indicates that Muar is ancestral to genotype IV and is a fifth genotype (Fig. 3b). This result supports the hypothesis that JEV originated in Indonesia/Malaysia. Furthermore, none of approximately 300 JEV sequences are closely related to strain Muar, indicating that this lineage might be extinct. The isolation of strain Muar from a human might indicate that all JEV strains, including genotype IV, can cause human disease.
West Nile virus
WNV, a member of the JEV serological and genetic group of the Flavivirus genus, was first isolated in 1937 in Uganda, and until recently was only found in the OLD WORLD. Febrile human disease caused by WNV was first reported in the 1950s in Israel. WNV and JEV have many common ecological features. The enzootic transmission cycle of WNV involves the transmission of virus among birds by Culex spp. In common with JEV52, birds are excellent amplifying hosts for WNV53, as they remain viraemic for several days, allowing migratory species to carry the virus over long distances. However, unlike JEV, pigs are not amplifying hosts. WNV also infects a wide variety of vertebrates through Culex spp., including dead-end hosts such as humans and equines. WNV can cause disease in these incidental hosts at a frequency of approximately 1 in 150 infections. Until recently, WNV was predominantly associated with febrile illness, only rarely causing encephalitis. However, in 1996, a large outbreak of human WNV infection, with a high incidence of encephalitis, was recorded in Romania. Subsequently, in 1998, the virus caused an epidemic in Russia that was characterized by many cases of neurological disease. These human outbreaks were accompanied by equine epizootics in Israel, France and Tunisia.
In 1999, WNV emerged in the New World when it was identified as the aetiological agent of an encephalitis outbreak in New York. The 62 documented human cases included 7 fatalities, and the 25 equine cases included 9 fatalities54. Since the year 2000, WNV has spread across North America and into Central America, with virus isolated in Mexico, El Salvador and the Caribbean Islands. In 2002, WNV caused the largest recorded outbreak of flavivirus encephalitis in the western hemisphere with a total of 4,156 human cases. Neurological symptoms were recorded in 2,942 cases, and there were 277 documented fatalities. There were also more than 15,000 reported equine cases during this outbreak. The epidemic continued in 2003, with the total number of human cases increasing to 9,858, of which 2,605 developed neurological disease. There were 262 recorded deaths. The number of WNV infections is therefore increasing, partly because of increased awareness and more frequent diagnosis of the disease. The number of cases of encephalitis was similar in 2002 and 2003, and the mortality rate remained constant at 10% (Ref. 55).
Phylogeny of WNV. Phylogenetic studies using both complete and partial genome sequences have shown that the WNV strain found in New York in 1999 is most closely related to a 1998 Israeli strain isolated from a goose, which indicates that the virus was recently introduced into the United States. This conclusion was supported by serological studies that showed a lack of anti-WNV antibodies in individuals living in New York prior to 1999 (Ref. 54).
Initial studies revealed that the nucleotide sequences of strains found on the eastern seaboard of the United States were similar to the prototype 'New York 1999' (NY99) strain isolated from a flamingo in the Bronx Zoo, and indicated that the NY99 strain spread westward in 2000—2002. Recent studies have shown that a genetic variant termed 'North America' has arisen since 2001 and has replaced NY99 as the dominant genotype present in North America56. The 'North America' genotype only differs by 0.18% from NY99, but has characteristic nucleotide and amino-acid substitutions. Other genetic variants have been identified, including a south-east coastal Texas genotype that differs by 0.27% from NY99 and by 0.55% from 'North America', and a Mexican genotype that differs by 0.45% from NY99 (Ref. 57). Overall, these data might indicate a lack of Darwinian selection in the evolution of a particular genetic variant, and support genetic drift of the virus during its dissemination across North America. Consistent with this hypothesis, the southeast coastal Texas genotype was only isolated in 2002 from the Texas—Louisiana border, and might have become extinct. To date, there is no evidence for selection of any phenotypic differences among North American strains, and it remains to be seen if WNV will evolve towards reduced virulence as it adapts to new hosts in the western-hemisphere ecosystem.
Emergence of WNV. There are many hypotheses about the mechanism of WNV emergence in the United States. It is unlikely that viraemic human travellers are a source of the virus, because humans only develop a low-titre viraemia and are therefore considered dead-end hosts. Bioterrorism is also an unlikely source, as the epicentre of the 1999 outbreak was in the Queen's district of New York City, and the virus was transmitted by mosquitoes. One explanation is that WNV was introduced to New York by a viraemic animal that was subsequently bitten by a mosquito, which then spread the disease to humans. Many exotic animal species, particularly birds, are imported into the United States through New York, and it is possible that an imported viraemic bird initiated transmission. Alternatively, an infected migratory bird might have carried the virus to the United States. Normally, migratory birds fly in a north—south direction, rather than in an east—west direction. The north—south route would imply that the virus entered the United States from Central/South America. This seems unlikely, as WNV was only found in the southern United States at least one year after it was discovered in New York. Another possibility is the east—west route. Although this might also seem unlikely, vagrant racing-pigeons from Europe occasionally appear in the United States. Recently, the UK media reported that a homing pigeon called 'Billy' was released in France en route to Britain, but instead flew to New York58. Finally, an infected mosquito that was transported aboard an aircraft could have introduced WNV to the United States. Aircraft departing from the Middle East to the United States are not routinely treated with insecticide, and mosquitoes often enter aeroplanes — attracted by humans or bright light.
Spread of WNV. Since the introduction of WNV into New York in 1999, studies have traced the virus as it spreads across North America. Many flaviviruses such as yellow fever virus (YFV) and DENV have a narrow host range consisting of a few arthropod and vertebrate species. By contrast, the host range of the North American WNV strain is enormous. To date, the virus can infect at least 49 species of mosquitoes and ticks, although transmission among these species has only been shown in a few cases. Similarly, at least 225 species of birds and at least 29 animals (including horses, cattle, llamas, alligators, cats, dogs, wolves and sheep) can become infected59,60. Animal species in both the Old and the New World have antibodies to WNV, but virus is rarely isolated from animals in the Old World compared with those in the New World. This is probably because WNV seldom causes clinically apparent disease in birds and vertebrates in the Old World. However, human and veterinary outbreaks that have occurred in Europe, the Middle East and north Africa61,62 during the past 10 years could indicate that recent isolates of WNV are more virulent or pathogenic than previous isolates. WNV seems to be highly 'promiscuous' in the New World compared with the Old World, demonstrating the dramatic effects of a new geographical range with non-immune hosts, which can result in the evolution of more virulent strains. The traditional mosquito-borne cycle still remains the main route of transmission of WNV in North America. However, differences in surveillance and the high incidence of human infection through blood transfusion, mother-to-foetus transmission, transmission in breast milk and by organ transplantation have caused major public health concerns63,64,65,66,67,68,69.
Adaptation of WNV to new environments. An important question that remains unanswered is how WNV will adapt to new ecosystems in the New World. There are two main hypotheses based on comparisons with other members of the JEV group members. First, WNV could become enzootic and endemic, similar to St Louis encephalitis virus (SLEV), and cause limited human disease through spillover. Alternatively, the virus could become epidemic, similar to Asian strains of JEV, and could cause annual outbreaks affecting large numbers of humans and animals. The 'explosive' nature of the North-American WNV epidemic as well as the equine and avian epizootics might reflect highly efficient enzootic amplification and avian virulence. North-American birds have probably not developed resistance to WNV because of low levels of viral exposure. Virulence in the avian host might subsequently decline because of the strong selection for resistance to WNV resulting from high bird-mortality rates. If resistance is accompanied by a decline in viraemia levels, enzootic amplification could subside. Then, spillover to humans and equines would diminish. The transport of WNV into Latin America by migratory birds raises important questions about pre-existing human and avian immunity to other flaviviruses such as SLEV, DENV, YFV and Rocio virus. Cross-reactive immunity could protect against WNV infection and/or disease, or could increase pathogenesis through immune enhancement, which is thought to contribute to the aetiology of dengue hemorrhagic fever. It is still not known if WNV and South American flaviviruses can share the same hosts and ecosystems (similar to WNV and SLEV in the United States). It is also not known how WNV will spread and cause human disease in the presence of flavivirus antibodies.
Competition between WNV and SLEV for avian hosts and mosquito vectors might be expected to result in competitive exclusion of one of these viruses70. However, these viruses coexist in the southern United States where they seem to use the same avian hosts and vectors. Low rates of immunity to SLEV in birds and low rates of infection in C. quinquefasciatus might indicate that host resources are not limiting factors, which could allow for indefinite coexistence.
Dengue virus
DENVs (serotypes 1—4) are the most important arbovirus human pathogens, and are also unusual as they use humans as reservoir hosts (see further information in the online links box). During the past 50 years, the prevalence of dengue fever, as well as life-threatening dengue haemorrhagic fever and shock syndromes, has increased exponentially, with approximately 2,500 million people (two-fifths of the world's population) at risk, and about 50 million cases recorded each year71. In urban settings, DENVs are transmitted among human hosts by the PERIDOMESTIC mosquito vectors Aedes aegypti and Aedes albopictus. However, studies of dengue virus ecology in sylvatic habitats of west Africa72 and Malaysia73,74 have identified transmission cycles involving non-human primates as reservoir hosts and arboreal, tree-hole dwelling Aedes (Stegomyia) spp. as vectors. Efficient inter-human DENV transmission probably requires a human population of 10,000 to 1 million people, a feature of urban civilizations that did not exist until about 4,000 years ago, and therefore the sylvatic cycle is probably ancestral75. Endemic/epidemic DENV is therefore thought to have evolved in Africa or Asia from sylvatic viral forms. Initial phylogenetic studies of both endemic/epidemic and sylvatic strains of DENV-2 showed evolutionary divergence of these ecologically distinct forms76. Recent studies indicate that epidemic/endemic forms of DENV-1, -2 and -4, which now use humans as reservoir hosts, evolved independently from sylvatic progenitors in the past 2,000 years, accompanied by host-range changes from non-human primates to humans, and from arboreal Aedes spp. to A. aegypti and A. albopictus vectors3,77 (Fig. 4). The highly efficient peridomestic transmission cycle — which is now independent of the ancestral sylvatic cycles — benefits greatly from the ecology of A. aegypti. This species lays its eggs in water-storage containers and in refuse, it readily enters human habitations and it often takes several blood meals during each reproductive cycle for both egg production and nutrition. Once infected, this competent vector transmits the virus to many human hosts78,79. Interestingly, as with sylvatic DENV, A. aegypti originated from another tree-hole-dwelling Aedes mosquito which is found in sylvatic African habitats (Aedes aegypti formosus)80. However, the sylvatic African forms of DENV do not use A. aegypti formosus as a principal vector, and this subspecies is relatively refractory to infection81. Yellow fever virus also uses A. aegypti aegypti as its main vector during African urban epidemics, but this cycle seems to be temporary and cannot be detected during inter-epidemic periods. It is not known why YFV has not adapted to a permanent human—A. aegypti endemic cycle, which would have devastating public health implications.

Phylogenetic tree derived from envelope-protein gene sequences (1,512 nucleotides) using neighbour-joining analysis. Endemic strains isolated from humans or peridomestic vectors are shown in purple and sylvatic strains isolated from non-human primates or arboreal mosquitoes are shown in green. Because the phenotypic change from sylvatic to endemic transmission could have occurred at any point along the indicated branches (shown by pink arrow boxes), time estimates for dengue emergence are represented by maximal values plus one standard deviation (derived from synonymous substitution rate estimates). DENV, dengue virus. Reproduced with permission from Ref. 3© (2000) American Society for Microbiology.
The adaptation of RNA viruses to new hosts is generally host-specific28. The evolutionary hypothesis therefore predicts that A. aegypti and A. albopictus should have increased susceptibility for endemic/epidemic DENV compared with its sylvatic progenitors. Recent experimental infection studies of populations of A. aegypti and A. albopictus support this hypothesis. When fed blood meals containing equivalent viral titres, the endemic/epidemic strains of DENV-2 consistently infect a higher proportion of mosquitoes than sylvatic strains82. If similar adaptation to human hosts occurred during DENV evolution, differences in pathogenicity might be expected between the endemic and sylvatic DENV strains. Studies are required in regions of Africa and Asia, where humans are exposed to sylvatic infection, to confirm these differences. Genetic studies are required to quantify the number of mutations leading to vector adaptation during emergence of urban dengue from ancestral sylvatic cycles. This will allow assessment of the frequency with which arboviruses can undergo host adaptation. These studies are important because DENV vaccines are currently being developed83 that could eradicate urban cycles that rely on humans as exclusive reservoir hosts. (The smallpox virus has been eradicated from natural transmission by smallpox vaccine and, similarly, poliovirus is nearly eliminated worldwide.) However, any eradication programme would rely on an inability of the sylvatic strains to re-emerge — currently the likelihood of this is not known.
Experimental model systems for arboviruses
Although host-range changes have occurred throughout arbovirus evolution, with important consequences for human health, the ease and frequency with which these events occur is unknown. Viruses that have obligate alternating replication cycles in taxonomically disparate hosts — vertebrate and arthropod — should be compromised because they are 'generalists', owing to their fitness in both hosts. Other viruses that use a single host as their main reservoir might be able to adapt more readily to related hosts. This might account for the slow rates of evolution of most arboviruses (approximately 104 substitutions per nucleotide per year) compared with many single-host RNA viruses (approximately 102—103 substitutions per nucleotide per year)84. Experimental cell-culture model systems have been developed to test this hypothesis, but results are only partially supportive. In vitro transmission cycles of EEEV have been established by serial passage in vertebrate (baby-hamster kidney), or mosquito (A. albopictus C6/36) cells85. EEEV has also been introduced into an alternating-host cycle of both cell types to mimic the natural transmission cycle. As predicted, virus-fitness increases occurred in both hosts following specialization. Viruses that had specialized to one host type showed a decline in fitness in the alternate cells, as well as in comparable vertebrate or mosquito cells, consistent with specificity of adaptation during specialization. Surprisingly, the alternating-host-cell cycles resulted in simultaneous adaptation to both cell lines, with increased levels of fitness comparable to those associated with host specialization, contradicting the hypothesis that arboviruses are limited in their adaptation potential by the alternating-host transmission cycle. However, specialization resulted in a larger number of mutations than alternating-host passages, consistent with the reduced rates of nucleotide-sequence changes typical of arboviruses. Alternating passage of EEEV in chicken and mosquito cell lines produced similar results86. Another arbovirus, vesicular stomatitis virus (VSV), can also adapt simultaneously to sandfly and vertebrate host cells, and the mutation rate of the virus in specialized compared to alternating-host passages is the same. This contradicts the hypothesis that arboviruses are constrained in their evolutionary potential87. Surprisingly, VSV populations that are specialized for replication in vertebrate cells showed fitness-increases for replication in sandfly cells, indicating a lack of specificity in some adaptation events. Further study of these model systems is needed to determine if alternating passages generate mutations that increase fitness in both cell types, or if they generate a polymorphic population with host-cell-specific adaptive mutations.
Recent in vivo experiments have produced results that contradict the cell-culture experimental models of arbovirus evolution. When VEEV was placed into a laboratory transmission cycle — consisting of ten cycles in hamsters alternating with ten cycles in A. aegypti mosquitoes — genetic stability of the virus isolates was observed, and virus fitness was either reduced or unchanged. However, specialization for replication in the hamster host resulted in rapid increases in virus fitness with many mutations, consistent with virus adaptation and evolutionary constraints imposed by the alternating-host cycle of arboviruses88. Cell-culture models probably do not fully reflect in vivo conditions, as artefacts (such as selection for virus binding to heparin sulphate) might distort results89,90.
The development of experimental systems to study mechanisms of replication of these important human and animal pathogens in different hosts should reveal how these RNA viruses emerge to cause disease in humans. Although it is still unclear if there are common mechanisms of emergence of arboviruses, studies using molecular genetics and viral ecology should enable researchers to predict emergent strains. Furthermore, common determinants of emergence could be used to develop rationally designed antiviral strategies.


References
Gao, F. et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 397, 436—441 (1999).
Korber, B. et al. Timing the ancestor of the HIV-1 pandemic strains. Science 288, 1789—1796 (2000).
Wang, E. et al. Evolutionary relationships of endemic/epidemic and sylvatic dengue viruses. J. Virol. 74, 3227—3234 (2000). This paper demonstrates the convergent evolution of endemic DENV virus strains from three of the four serotpes of sylvatic progenitors, and places a time frame on urban dengue emergence that is congruent with historical and epidemiological predictions.
Gubler, D. J. The global emergence/resurgence of arboviral diseases as public health problems. Arch. Med. Res. 33, 330—342 (2002).
Marra, M. A. et al. The genome sequence of the SARS-associated coronavirus. Science 300, 1399—1404 (2003).
Guan, Y. et al. Isolation and characterization of viruses related to the SARS coronavirus from animals in southern China. Science 302, 276—278 (2003).
Rota, P. A. et al. Characterization of a novel coronavirus associated with severe acute respiratory syndrome. Science 300, 1394—1399 (2003).
Rosenbloom, M., Leikin, J. B., Vogel, S. N. & Chaudry, Z. A. Biological and chemical agents: a brief synopsis. Am. J. Ther. 9, 5—14 (2002).
Kubes, V. & Rios, F. A. The causative agent of infectious equine encephalomyelitis in Venezuela. Science 90, 20—21 (1939).
Beck, C. E. & Wyckoff, R. W. G. Venezuelan equine encephalomyelitis. Science 88, 530 (1938).
Weaver, S. C., Ferro, C., Barrera, R., Boshell, J. & Navarro, J. Venezuelan equine encephalitis. Annu. Rev. Entomol. 49, 141—174 (2004).
Walton, T. E. & Grayson, M. A. in The Arboviruses: Epidemiology and Ecology Vol. IV (ed. Monath, T. P.) 203—231 (CRC, Boca Raton, Florida, 1988).
Walton, T. E., Alvarez, O., Buckwalter, R. M. & Johnson, K. M. Experimental infection of horses with enzootic and epizootic strains of Venezuelan equine encephalomyelitis virus. J. Infect. Dis. 128, 271—282 (1973).
Wang, E. et al. Virulence and viremia characteristics of 1992 epizootic subtype IC Venezuelan equine encephalitis viruses and closely related enzootic subtype ID strains. Am. J. Trop. Med. Hyg. 65, 64—69 (2001).
Rico-Hesse, R., Weaver, S. C., de Siger, J., Medina, G. & Salas, R. A. Emergence of a new epidemic/epizootic Venezuelan equine encephalitis virus in South America. Proc. Natl Acad. Sci. USA 92, 5278—5281 (1995).
Weaver, S. C. et al. Re-emergence of epidemic Venezuelan equine encephalomyelitis in South America. VEE Study Group. Lancet 348, 436—440 (1996).
Oberste, M. S. et al. Association of Venezuelan equine encephalitis virus subtype IE with two equine epizootics in Mexico. Am. J. Trop. Med. Hyg. 59, 100—107 (1998).
Zarate, M. L., Scherer, W. F. & Dickerman, R. W. El virus de la encephalitis equina de Venezuela como determinante de infecciones en humanos, descripción de un caso fatal ocurrido en Jaltipán Veracruz en 1965. Rev. Invest. Salud Publica 30, 296—302 (1965).
Johnson, K. M. & Martin, D. H. Venezuelan equine encephalitis. Adv. Vet. Sci. Comp. Med. 18, 79—116 (1974).
Young, N. A. & Johnson, K. M. Antigenic variants of Venezuelan equine encephalitis virus: their geographic distribution and epidemiologic significance. Am. J. Epidemiol. 89, 286—307 (1969). This landmark paper established the antigenic relationships of enzootic and epizootic strains of VEEVs and provided the framework for future genetic studies that identified the origins of outbreaks.
Rico-Hesse, R., Roehrig, J. T., Trent, D. W. & Dickerman, R. W. Genetic variation of Venezuelan equine encephalitis virus strains of the ID variety in Colombia. Am. J. Trop. Med. Hyg. 38, 195—204 (1988).
Kinney, R. M., Tsuchiya, K. R., Sneider, J. M. & Trent, D. W. Genetic evidence that epizootic Venezuelan equine encephalitis (VEE) viruses may have evolved from enzootic VEE subtype I-D virus. Virology 191, 569—580 (1992).
Weaver, S. C., Bellew, L. A. & Rico-Hesse, R. Phylogenetic analysis of alphaviruses in the Venezuelan equine encephalitis complex and identification of the source of epizootic viruses. Virology 191, 282—290 (1992). This paper provides the first phylogenetic evidence that epizootic strains of VEEV have evolved repeatedly from enzootic subtype ID progenitors, and correctly predicts the occurrence of additional outbreaks after a 19-year absence.
Brault, A. C. et al. Potential sources of the 1995 Venezuelan equine encephalitis subtype IC epidemic. J. Virol. 75, 5823—5832 (2001).
Wang, E. et al. Genetic and phenotypic changes accompanying the emergence of epizootic subtype IC Venezuelan equine encephalitis viruses from an enzootic subtype ID progenitor. J. Virol. 73, 4266—4271 (1999).
Johnson, B. J., Brubaker, J. R., Roehrig, J. T. & Trent, D. W. Variants of Venezuelan equine encephalitis virus that resist neutralization define a domain of the E2 glycoprotein. Virology 177, 676—683 (1990).
Brault, A. C., Powers, A. M., Holmes, E. C., Woelk, C. H. & Weaver, S. C. Positively charged amino acid substitutions in the E2 envelope glycoprotein are associated with the emergence of Venezuelan equine encephalitis virus. J. Virol. 76, 1718—1730 (2002).
Holland, J. & Domingo, E. Origin and evolution of viruses. Virus Genes 16, 13—21 (1998).
Oberste, M. S., Schmura, S. M., Weaver, S. C. & Smith, J. F. Geographic distribution of Venezuelan equine encephalitis virus subtype IE genotypes in Central America and Mexico. Am. J. Trop. Med. Hyg. 60, 630—634 (1999).
Gonzalez-Salazar, D., Estrada-Franco, J. G., Carrara, A. S., Aronson, J. F. & Weaver, S. C. Equine amplification and virulence of subtype IE Venezuelan equine encephalitis viruses isolated during the 1993 and 1996 Mexican epizootics. Emerg. Infect. Dis. 9, 161—168 (2003).
Brault, A. C. et al. Venezuelan equine encephalitis emergence: Enhanced vector infection from a single amino acid substitution in the envelope glycoprotein. Proc. Natl Acad. Sci. USA 101, 11344—11349 (2004).
Ferro, C. et al. Natural enzootic vectors of Venezuelan equine encephalitis virus, Magdalena Valley, Colombia. Emerg. Infect. Dis. 9, 49—54 (2003).
Linthicum, K. J. et al. Climate and satellite indicators to forecast Rift Valley fever epidemics in Kenya. Science 285, 397—400 (1999).
Kramer, L. D. & Scherer, W. F. Vector competence of mosquitoes as a marker to distinguish Central American and Mexican epizootic from enzootic strains of Venezuelan encephalitis virus. Am. J. Trop. Med. Hyg. 25, 336—346 (1976).
Brault, A. C., Powers, A. M. & Weaver, S. C. Vector infection determinants of Venezuelan equine encephalitis virus reside within the E2 envelope glycoprotein. J. Virol. 76, 6387—6392 (2002).
Bowen, G. S. & Calisher, C. H. Virological and serological studies of Venezuelan equine encephalomyelitis in humans. J. Clin. Microbiol. 4, 22—27 (1976).
Weaver, S. C. in The Encyclopedia of Arthropod-transmitted Infections (ed. Service, M. W.) 151—159 (CAB International, Wallingford, UK, 2001).
Reisen, W. K. in The Encyclopedia of Arthropod-transmitted Infections (ed. Service, M. W.) 558—563 (CAB International, Wallingford, UK, 2001).
Hahn, C. S., Lustig, S., Strauss, E. G. & Strauss, J. H. Western equine encephalitis virus is a recombinant virus. Proc. Natl Acad. Sci. USA 85, 5997—6001 (1988).
Hiroyama, T. Epidemiology of Japanese encephalitis (in Japanese). Saishin-Igaku 17, 1272—1280 (1962).
Mitamura, T., Kitaoka, M., Mori, K. & Okuba, K. Isolation of Japanese epidemic encephalitis from mosquitoes caught in nature. Tokyo Iji Shinshi 62, 820—831 (1938).
Scherer, W. F. Ecological studies of Japanese encephalitis in Japan, parts I—IX. Am. J. Trop. Med. Hyg. 8, 644—722 (1959). This paper is one of a series of ten papers that describes the ecology, epidemiology and transmission cycles of JEV.
Tan, R., Nalim, S., Suwasono, H. & Jennings, G. B. Japanese encephalitis virus isolated from seven species of mosquitoes collected at Semarang Regency, Central Java. Bul. Penelit. Kesehatan 21, 1—5 (1993).
Chen, W. R., Tesh, R. B. & Rico-Hesse, R. Genetic variation of Japanese encephalitis virus in nature. J. Gen. Virol. 71, 2915—2922 (1990).
Chen, W. R., Rico-Hesse, R. & Tesh, R. B. A new genotype of Japanese encephalitis virus from Indonesia. Am. J. Trop. Med. Hyg. 47, 61—69 (1992).
Kobayashi, Y., Hasegawa, H. & Yamauchi, T. Studies on the antigenic structure of Japanese encephalitis virus using monoclonal antibodies. Microbiol. Immunol. 29, 1069—1082 (1985).
Sumiyoshi, H. et al. Complete nucleotide sequence of the Japanese encephalitis virus genome RNA. Virology 161, 497—510 (1987).
Gould, E. A., de Lamballerie, X., Zanotto, P. M. & Holmes, E. C. Origins, evolution, and vector/host coadaptations within the genus Flavivirus. Adv. Virus Res. 59, 277—314 (2003).
Solomon, T. et al. Origin and evolution of Japanese encephalitis virus in southeast Asia. J. Virol. 77, 3091—3098 (2003). This paper proposes that JEV originated in the Indonesia and Malaysia regions and has since spread to other parts of Asia and Australasia.
Hancock, J. & Kushlan, J. The Herons Handbook (Harper and Row, New York, 1984).
Hasegawa, H., Yoshida, M., Fujita, S. & Kobayashi, Y. Comparison of structural proteins among antigenically different Japanese encephalitis virus strains. Vaccine 12, 841—844 (1994).
Scherer, W. F., Buescher, E. L. & McClure, H. E. Ecologic studies of Japanese encephalitis virus in Japan. V. Avian factors. Am. J. Trop. Med. Hyg. 8, 689—697 (1959).
Work, T. H., Hurlbut, H. S. & Taylor, R. M. Indigenous wild birds of the Nile Delta as potential West Nile virus circulating reservoirs. Am. J. Trop. Med. Hyg. 4, 872—888 (1955).
Lanciotti, R. S. et al. Origin of the West Nile virus responsible for an outbreak of encephalitis in the northeastern United States. Science 286, 2333—2337 (1999). This paper describes the emergence of WNV in the United States.
O'Leary, D. R. et al. The epidemic of West Nile virus in the United States, 2002. Vector Borne Zoonotic Dis. 4, 61—70 (2004).
Davis, C. T. et al. Genetic variation among geographically distinct West Nile virus isolates collected in the United States during 2002. Emerg. Infect. Dis. 9, 1423—1429 (2003).
Estrada-Franco, J. G. et al. West Nile virus in Mexico: evidence of widespread circulation since July 2002. Emerg. Infect. Dis. 9, 1604—1607 (2003).
Finney, S. Hopelessly lost, the homing pigeon whose cross-channel hop took him to New York. Daily Mail (Lond.) (23 June 2003).
Marra, P. P., Griffing, S. M. & McLean, R. G. West Nile virus and wildlife health. Emerg. Infect. Dis. 9, 898—899 (2003).
McLean, R. G. West Nile Virus: emerging threat to public health and animal health. J. Vet. Med. Educ. 30, 143—144 (2003).
Malkinson, M. & Banet, C. The role of birds in the ecology of West Nile virus in Europe and Africa. Curr. Top. Microbiol. Immunol. 267, 309—322 (2002).
Murgue, B., Zeller, H. & Deubel, V. The ecology and epidemiology of West Nile virus in Africa, Europe and Asia. Curr. Top. Microbiol. Immunol. 267, 195—221 (2002).
Intrauterine West Nile virus infection — New York, 2002. MMWR Morb. Mortal Wkly. Rep. 51, 1135—1136 (2002).
Detection, of West Nile virus in blood donations — United States, 2003. MMWR Morb. Mortal Wkly Rep. 52, 769—772 (2003).
Petersen, L. R., Roehrig, J. T. & Hughes, J. M. West Nile virus encephalitis. N. Engl. J. Med. 347, 1225—1226 (2002).
Pealer, L. N. et al. Transmission of West Nile virus through blood transfusion in the United States in 2002. N. Engl. J. Med. 349, 1236—1245 (2003).
Iwamoto, M. et al. Transmission of West Nile virus from an organ donor to four transplant recipients. N. Engl. J. Med. 348, 2196—203 (2003).
Petersen, L. R., Marfin, A. A. & Gubler, D. J. West Nile virus. JAMA 290, 524—528 (2003).
Harrington, T. et al. West Nile virus infection transmitted by blood transfusion. Transfusion 43, 1018—1022 (2003).
Gause, G. F. The Struggle for Existence (Dover Publications, New York, 1971).
WHO. Dengue and dengue haemorrhagic fever. [online], <http://www.who.int/mediacentre/factsheets/fs117> (April, 2002).
Diallo, M. et al. Amplification of the sylvatic cycle of dengue virus type 2, Senegal, 1999—2000: entomologic findings and epidemiologic considerations. Emerg. Infect. Dis. 9, 362—367 (2003).
Rudnick, A. Studies of the ecology of dengue in Malaysia: a preliminary report. J. Med. Entomol. 2, 203—208 (1965).
Rudnick, A. in Proceedings of the International Conference on Dengue/DHF (eds Pang, T. & Pathmanathan, R.) 7 (University of Malaysia Press, Kuala Lumpur, 1984).
Gubler, D. J. in Dengue and Dengue Hemorrhagic Fever (eds Gubler, D. J. & Kuno, G.) 1—22 (CAB International, New York, 1997).
Rico-Hesse, R. Molecular evolution and distribution of dengue viruses type 1 and 2 in nature. Virology 174, 479—493 (1990).
Holmes, E. C. & Twiddy, S. S. The origin, emergence and evolutionary genetics of dengue virus. Infect. Genet. Evol. 3, 19—28 (2003).
Harrington, L. C., Edman, J. D. & Scott, T. W. Why do female Aedes aegypti (Diptera: Culicidae) feed preferentially and frequently on human blood? J. Med. Entomol. 38, 411—422 (2001).
Weaver, S. C., Coffey, L. L., Nussenzveig, R., Ortiz, D. & Smith, D. in Microbe—Vector Interactions in Vector-borne Diseases (eds Gillespie, S. H., Smith, G. L. & Osbourn, A.) 139—180 (Cambridge Univ. Press, Cambridge, 2004).
Tabachnick, W. J. & Powell, J. R. A world-wide survey of genetic variation in the yellow fever mosquito, Aedes aegypti. Genet. Res. 34, 215—229 (1979).
Black, W. C. et al. Flavivirus susceptibility in Aedes aegypti. Arch. Med. Res. 33, 379—88 (2002).
Moncayo, A. C. et al. Emergence of epidemic dengue through the adaptation of sylvatic progenitor viruses to anthropophilic mosquito vectors. Emerg. Infect. Dis. (in the press).
Guirakhoo, F. et al. Viremia and immunogenicity in nonhuman primates of a tetravalent yellow fever—dengue chimeric vaccine: genetic reconstructions, dose adjustment, and antibody responses against wild-type dengue virus isolates. Virology 298, 146—159 (2002).
Weaver, S. C., Rico-Hesse, R. & Scott, T. W. Genetic diversity and slow rates of evolution in New World alphaviruses. Curr. Top. Microbiol. Immunol. 176, 99—117 (1992).
Weaver, S. C., Brault, A. C., Kang, W. & Holland, J. J. Genetic and fitness changes accompanying adaptation of an arbovirus to vertebrate and invertebrate cells. J. Virol. 73, 4316—4326 (1999).
Cooper, L. A. & Scott, T. W. Differential evolution of eastern equine encephalitis virus populations in response to host cell type. Genetics 157, 1403—1412 (2001).
Novella, I. S., Hershey, C. L., Escarmis, C., Domingo, E. & Holland, J. J. Lack of evolutionary stasis during alternating replication of an arbovirus in insect and mammalian cells. J. Mol. Biol. 287, 459—465 (1999).
Brault, A. C. Genetic Analysis of Epizootic Venezuelan Equine Encephalitis Virus Emergence Mechanisms. Thesis, Univ. Texas Medical Branch, (2001).
Byrnes, A. P. & Griffin, D. E. Binding of Sindbis virus to cell surface heparan sulfate. J. Virol. 72, 7349—7356 (1998).
Klimstra, W. B., Ryman, K. D. & Johnston, R. E. Adaptation of Sindbis virus to BHK cells selects for use of heparan sulfate as an attachment receptor. J. Virol. 72, 7357—7366 (1998).
Weaver, S. C. et al. Genetic determinants of Venezuelan equine encephalitis emergence. Arch. Virol. Suppl. 18, 43—64 (2004).
Weaver, S. C., Ferro, C., Barrera, R., Boshell, J. & Navarro, J. C. Venezuelan equine encephalitis. Annu. Rev. Entomol. 49, 141—174 (2004).
Acknowledgements
S.C.W. is suppported by the National Institutes of Health. A.D.T.B. is supported by the State of Texas Higher Education Coordinating Board and the Centers for Diseases Control.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Related links
Related links
DATABASES
Entrez
Infectious Disease Database
FURTHER INFORMATION
Glossary
- EPIZOOTIC
-
Higher than average amplification, or occurrence, of a disease or pathogen in non-human animals.
- STRAINS
-
Individual isolates or variants of a virus.
- VIRAEMIA
-
Presence of virus in the bloodstream — usually essential for transmission of arboviruses by arthropod vectors.
- SYLVATIC
-
Occurring in forest habitats.
- ENZOOTIC
-
A disease or maintenance transmission cycle occurring continuously among non-human animals in a particular region or locality.
- SUBTYPE
-
A distinct virus variant that can be antigenically distinguished from other closely related variants.
- SYMPATRIC
-
Having overlapping geographical distributions.
- ZOONOTIC
-
Pathogens or diseases that normally circulate among non-human animals but that can be transmitted to humans.
- OLD WORLD
-
Those parts of the world known to Europeans before the voyages of Christopher Columbus — Europe, Asia and Africa. The New World refers to the American continents.
- PERIDOMESTIC
-
In, and around, human habitations.
Rights and permissions
About this article
Cite this article
Weaver, S., Barrett, A. Transmission cycles, host range, evolution and emergence of arboviral disease. Nat Rev Microbiol 2, 789–801 (2004). https://doi.org/10.1038/nrmicro1006
Issue Date:
DOI: https://doi.org/10.1038/nrmicro1006
This article is cited by
-
Scoping review of Culex mosquito life history trait heterogeneity in response to temperature
Parasites & Vectors (2023)
-
Studies on the antiviral activity of chebulinic acid against dengue and chikungunya viruses and in silico investigation of its mechanism of inhibition
Scientific Reports (2022)
-
A brief review on novel biomarkers identified and advanced biosensing technologies developed for rapid diagnosis of Japanese Encephalitis Virus
Proceedings of the Indian National Science Academy (2022)
-
Using Environmental Sampling to Enable Zoonotic Pandemic Preparedness
Journal of the Indian Institute of Science (2022)
-
Divide-and-conquer: machine-learning integrates mammalian and viral traits with network features to predict virus-mammal associations
Nature Communications (2021)
