
Key Points
-
Alteration of chromatin architecture by means of post-translational modifications of histone tails is an important process for the regulation of gene expression. The coordinated actions of histone-tail acetylation, methylation and phosphorylation, and ATP-dependent chromatin remodelling, allow fine control of gene activation or repression.
-
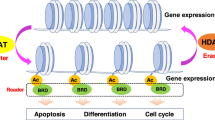
Histone acetylation and deacetylation is regulated by the opposing activities of histone acetyltransferases (HATs) and histone deacetylatransferases (HDACs).
-
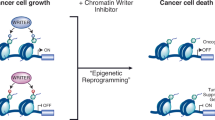
In cancer, the molecular processes that lead to inappropriate expression of genes due to altered chromatin structure are now being identified, and aberrant acetylation of histone tails is strongly linked to carcinogenesis. So, targeting the transcriptional lesions that lead to neoplasia provides an opportunity for therapeutic intervention at the very apex of the transformation process. Such therapies could affect several molecular programmes, and would therefore be more powerful than targeting the end stages of a single disrupted molecular pathway.
-
HDAC inhibitors are an exciting new class of chemotherapeutic drugs. These agents interact with the catalytic site of HDACs, block substrate access and allow hyperacetylation of histone tails.
-
The anticancer potential of HDAC inhibitors stems from their ability to affect several cellular processes that are dysregulated in neoplastic cells. Principally, activation of differentiation programmes, inhibition of the cell cycle and induction of apoptosis are the key antitumour activities of HDAC inhibitors. In addition, activation of the host immune response and inhibition of angiogenesis might also have important roles in HDAC-inhibitor-mediated tumour regression in vivo.
-
Much interest and excitement has been generated following the success of HDAC inhibitors in potently inhibiting tumour progression in rodent models. HDAC inhibitors can mediate histone acetylation in vivo, induce tumour-cell differentiation or apoptosis depending on the cell type, and are associated with minimal toxicity as assessed by weight loss and post-mortem analyses.
-
Given the success of HDAC inhibitors in preclinical studies, Phase I and II clinical trials using several different inhibitors have now been initiated. These drugs seem to be well tolerated at the doses required to hyperacetylate histones and achieve clinical outcomes, and their use in combination therapies is an area that can be further exploited in the clinic.
Abstract
The opposing actions of histone acetyltransferases (HATs) and histone deacetylases (HDACs) allow gene expression to be exquisitely regulated through chromatin remodelling. Aberrant transcription due to altered expression or mutation of genes that encode HATs, HDACs or their binding partners, is a key event in the onset and progression of cancer. HDAC inhibitors can reactivate gene expression and inhibit the growth and survival of tumour cells. The remarkable tumour specificity of these compounds, and their potency in vitro and in vivo, underscore the potential of HDAC inhibitors as exciting new agents for the treatment of cancer.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 12 print issues and online access
$209.00 per year
only $17.42 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout



Similar content being viewed by others
References
Hanahan, D. & Weinberg, R. A. The hallmarks of cancer. Cell 100, 57–70 (2000).An excellent review, which outlines the molecular defects that must occur for the transformation of a normal cell into a tumour cell.
Jacobson, S. & Pillus, L. Modifying chromatin and concepts of cancer. Curr. Opin. Genet. Dev. 9, 175–184 (1999).
Wu, J. & Grunstein, M. 25 Years after the nucleosome model: chromatin modifications. Trends Biochem. Sci. 25, 619–623 (2000).
Luger, K., Mader, A. W., Richmond, R. K., Sargent, D. F. & Richmond, T. J. Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature 389, 251–260 (1997).The nucleosome crystal structure was solved in this study.
Jenuwein, T. & Allis, C. D. Translating the histone code. Science 293, 1074–1080 (2001).
Flaus, A. & Owen-Hughes, T. Mechanisms for ATP-dependent chromatin remodelling. Curr. Opin. Genet. Dev. 11, 148–154 (2001).
Marmorstein, R. & Roth, S. Y. Histone acetyltransferases: function, structure, and catalysis. Curr. Opin. Genet. Dev. 11, 155–161 (2001).
Sterner, D. E. & Berger, S. L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 64, 435–459 (2000).
Gray, S. G. & Ekstrom, T. J. The human histone deacetylase family. Exp. Cell Res. 262, 75–83 (2001).
Khochbin, S., Verdel, A., Lemercier, C. & Seigneurin-Berny, D. Functional significance of histone deacetylase diversity. Curr. Opin. Genet Dev. 11, 162–166 (2001).
Fischle, W., Kiermer, V., Dequiedt, F. & Verdin, E. The emerging role of class II histone deacetylases. Biochem. Cell Biol. 79, 337–348 (2001).
Lee, H. J., Chun, M. & Kandror, K. V. Tip60 and HDAC7 interact with the endothelin receptor A and may be involved in downstream signaling. J. Biol. Chem. 276, 16597–16600 (2001).
Imai, S., Armstrong, C. M., Kaeberlein, M. & Guarente, L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403, 795–800 (2000).
Luo, J. et al. Negative control of p53 by Sir2α promotes cell survival under stress. Cell 107, 137–148 (2001).
Vaziri, H. et al. hSIR2(SIRT1) Functions as an NAD-dependent p53 deacetylase. Cell 107, 149–159 (2001).
Robertson, K. D. DNA methylation, methyltransferases, and cancer. Oncogene 20, 3139–3155 (2001).
Esteller, M. & Herman, J. G. Cancer as an epigenetic disease: DNA methylation and chromatin alterations in human tumours. J. Pathol. 196, 1–7 (2002)
Kouzarides, T. Histone acetylases and deacetylases in cell proliferation. Curr. Opin. Genet. Dev. 9, 40–48 (1999).
Muller, C. & Leutz, A. Chromatin remodeling in development and differentiation. Curr. Opin. Genet. Dev. 11, 167–174 (2001).
Muraoka, M. et al. p300 Gene alterations in colorectal and gastric carcinomas. Oncogene 12, 1565–1569 (1996).
Giles, R. H., Peters, D. J. & Breuning, M. H. Conjunction dysfunction: CBP/p300 in human disease. Trends Genet. 14, 178–183 (1998).
Gayther, S. A. et al. Mutations truncating the EP300 acetylase in human cancers. Nature Genet. 24, 300–303 (2000).
Goodman, R. H. & Smolik, S. CBP/p300 in cell growth, transformation, and development. Genes Dev. 14, 1553–1577 (2000).
Grossman, S. R. p300/CBP/p53 Interaction and regulation of the p53 response. Eur. J. Biochem. 268, 2773–2778 (2001).
Liang, J., Prouty, L., Williams, B. J., Dayton, M. A. & Blanchard, K. L. Acute mixed lineage leukemia with an inv(8)(p11q13) resulting in fusion of the genes for MOZ and TIF2. Blood 92, 2118–2122 (1998).
Panagopoulos, I. et al. Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16)(q22;p13). Hum. Mol. Genet. 10, 395–404 (2001).
Lin, R. J., Sternsdorf, T., Tini, M. & Evans, R. M. Transcriptional regulation in acute promyelocytic leukemia. Oncogene 20, 7204–7215 (2001).
Zelent, A., Guidez, F., Melnick, A., Waxman, S. & Licht, J. D. Translocations of the RARα gene in acute promyelocytic leukemia. Oncogene 20, 7186–7203 (2001).
Pandolfi, P. P. Transcription therapy for cancer. Oncogene 20, 3116–3127 (2001).
Heibert, S. W. et al. Mechanisms of transcriptional repression by the t(8;21)-, t(12;21)-, and inv(16)-encoded fusion proteins. Cancer Chemother. Pharmacol. 48, S31–S34 (2001).
Licht, J. D. AML1 and the AML1–ETO fusion protein in the pathogenesis of t(8;21) AML. Oncogene 20, 5660–5679 (2001).
Huynh, K. D., Fischle, W., Verdin, E. & Bardwell, V. J. BCoR, A novel corepressor involved in BCL-6 repression. Genes Dev. 14, 1810–1823 (2000).
McKinsey, T. A., Zhang, C. L. & Olson, E. N. Control of muscle development by dueling HATs and HDACs. Curr. Opin. Genet. Dev. 11, 497–504 (2001).
Soengas, M. S. et al. Inactivation of the apoptosis effector Apaf-1 in malignant melanoma. Nature 409, 207–211 (2001).
Teitz, T. et al. Caspase -8 is deleted or silenced preferentially in childhood neuroblastomas with amplification of MYCN. Nature Med. 6, 529–535 (2000).
Rountree, M. R., Bachman, K. E., Herman, J. G. & Baylin, S. B. DNA methylation, chromatin inheritance, and cancer. Oncogene 20, 3156–3165 (2001).
Dervan, P. B. & Burli, R. W. Sequence-specific DNA recognition by polyamides. Curr. Opin. Chem. Biol. 3, 688–693 (1999).
Mapp, A. K., Ansari, A. Z., Ptashne, M. & Dervan, P. B. Activation of gene expression by small molecule transcription factors. Proc. Natl Acad. Sci. USA 97, 3930–3935 (2000).
Liu, P. Q. et al. Regulation of an endogenous locus using a panel of designed zinc finger proteins targeted to accessible chromatin regions. Activation of vascular endothelial growth factor A. J. Biol Chem. 276, 11323–11334 (2001).References 38 and 39 describe the production of functional designer transcription factors using polymides and zinc-finger proteins.
Finnin, M. S. et al. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 401, 188–193 (1999).This group solved the structure of an HDAC inhibitor bound to an HDAC.
Marks, P. A., Richon, V. M., Breslow, R. & Rifkind, R. A. Histone deacetylase inhibitors as new cancer drugs. Curr. Opin. Oncol. 13, 477–483 (2001).
Weidle, U. H. & Grossmann, A. Inhibition of histone deacetylases: a new strategy to target epigenetic modifications for anticancer treatment. Anticancer Res. 20, 1471–1485 (2000).
Phiel, C. J. et al. Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem 276, 36734–36741 (2001).
Gottlicher, M. et al. Valproic acid defines a novel class of HDAC inhibitors inducing differentiation of transformed cells. EMBO J. 20, 6969–6978 (2001).References 43 and 44 describe the characterization of the antiepileptic drug valproic acid as an HDAC inhibitor.
Komatsu, Y. et al. Cyclic hydroxamic-acid-containing peptide 31, a potent synthetic histone deacetylase inhibitor with antitumor activity. Cancer Res. 61, 4459–4466 (2001).
Furumai, R. et al. Potent histone deacetylase inhibitors built from trichostatin A and cyclic tetrapeptide antibiotics including trapoxin. Proc. Natl Acad. Sci. USA 98, 87–92 (2001).
Mariadason, J. M., Corner, G. A. & Augenlicht, L. H. Genetic reprogramming in pathways of colonic cell maturation induced by short chain fatty acids: comparison with trichostatin A, sulindac, and curcumin and implications for chemoprevention of colon cancer. Cancer Res. 60, 4561–4572 (2000).The use of DNA microarrays to identify genes that are regulated by HDAC inhibitors.
Zhang, Y. & Reinberg, D. Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails. Genes Dev. 15, 2343–2360 (2001).
Cameron, E. E., Bachman, K. E., Myohanen, S., Herman, J. G. & Baylin, S. B. Synergy of demethylation and histone deacetylase inhibition in the re-expression of genes silenced in cancer. Nature Genet. 21, 103–107 (1999).This study shows the functional synergy between HDAC inhibitors and the DNA demethylating agent 5-azadC.
Marks, P. A., Richon, V. M. & Rifkind, R. A. Histone deacetylase inhibitors: inducers of differentiation or apoptosis of transformed cells. J. Natl Cancer Inst. 92, 1210–1216 (2000).
Marks, P. A. et al. Histone deacetylases and cancer: causes and therapies. Nature Rev. Cancer 1, 194–202 (2001)
Kramer, O. H., Gottlicher, M. & Heinzel, T. Histone deacetylase as a therapeutic target. Trends Endocrinol. Metab. 12, 294–300 (2001).
Qiu, L. et al. Histone deacetylase inhibitors trigger a G2 checkpoint in normal cells that is defective in tumor cells. Mol. Biol Cell. 11, 2069–2083 (2000).
Ruefli, A. A. et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of BID and production of reactive oxygen species. Proc. Natl Acad. Sci. USA 98, 10833–10838 (2001).The identification of a new mechanism of activation of the intrinsic apoptotic pathway by SAHA.
He, L. Z. et al. Histone deacetylase inhibitors induce remission in transgenic models of therapy-resistant acute promyelocytic leukemia. J. Clin. Invest. 108, 1321–1330 (2001).
Ferrara, F. F. et al. Histone deacetylase-targeted treatment restores retinoic acid signaling and differentiation in acute myeloid leukemia. Cancer Res. 61, 2–7 (2001).
Kosugi, H. et al. Histone deacetylase inhibitors are the potent inducer/enhancer of differentiation in acute myeloid leukemia: a new approach to anti-leukemia therapy. Leukemia 13, 1316–1324 (1999).
Lipinski, M. M. & Jacks, T. The retinoblastoma gene family in differentiation and development. Oncogene 18, 7873–7882 (1999).
Sandor, V. et al. p21-Dependent G1 arrest with downregulation of cyclin D1 and upregulation of cyclin E by the histone deacetylase inhibitor FR901228. Br. J. Cancer 83, 817–825 (2000).
Rosato, R. R., Wang, Z., Gopalkrishnan, R. V., Fisher, P. B. & Grant, S. Evidence of a functional role for the cyclin-dependent-kinase inhibitor p21WAF1/CIP1/MDA6 in promoting differentiation and preventing mitochondrial dysfunction and apoptosis induced by sodium butyrate in human myelomonocytic leukemia cells (U937). Int. J. Oncol. 19, 181–191 (2001).
Burgess, A. J. et al. Up-regulation of p21(WAF1/CIP1) by histone deacetylase inhibitors reduces their cytotoxicity. Mol. Pharmacol. 60, 828–837 (2001).
Vrana, J. A. et al. Induction of apoptosis in U937 human leukemia cells by suberoylanilide hydroxamic acid (SAHA) proceeds through pathways that are regulated by BCL-2/BCL-XL, c-JUN, and p21CIP1, but independent of p53. Oncogene 18, 7016–7025 (1999).
Zhu, L. & Skoultchi, A. I. Coordinating cell proliferation and differentiation. Curr. Opin. Genet. Dev. 11, 91–97 (2001).
Munster, P. N. et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid induces differentiation of human breast cancer cells. Cancer Res. 61, 8492–8497 (2001).
Kwon, S. H. et al. Apicidin, a histone deacetylase inhibitor, induces apoptosis and FAS/FAS ligand expression in human acute promyelocytic leukemia cells. J. Biol. Chem. 6, 6 (2001)
Glick, R. D. et al. Hybrid polar histone deacetylase inhibitor induces apoptosis and CD95/CD95 ligand expression in human neuroblastoma. Cancer Res. 59, 4392–4399 (1999).References 65 and 66 outline the potential role of the death-receptor pathway in apoptosis that is induced by HDAC inhibitors.
Mello, J. A. & Almouzni, G. The ins and outs of nucleosome assembly. Curr. Opin. Genet. Dev. 11, 136–141 (2001).
Green, D. R. Apoptotic pathways: paper wraps stone blunts scissors. Cell 102, 1–4 (2000).
Bonnotte, B. et al. Cancer cell sensitization to Fas-mediated apoptosis by sodium butyrate. Cell Death Differ. 5, 480–487 (1998).
Bernhard, D. et al. Inhibition of histone deacetylase activity enhances Fas receptor-mediated apoptosis in leukemic lymphoblasts. Cell Death Differ. 8, 1014–1021 (2001).
Wang, R., Brunner, T., Zhang, L. & Shi, Y. Fungal metabolite FR901228 inhibits c-Myc and Fas ligand expression. Oncogene 17, 1503–1508 (1998).
Bernhard, D. et al. Apoptosis induced by the histone deacetylase inhibitor sodium butyrate in human leukemic lymphoblasts. FASEB J. 13, 1991–2001 (1999).
Suzuki, T. et al. Effect of trichostatin A on cell growth and expression of cell cycle- and apoptosis-related molecules in human gastric and oral carcinoma cell lines. Int. J. Cancer 88, 992–997 (2000).
Cao, X. X., Mohuiddin, I., Ece, F., McConkey, D. J. & Smythe, W. R. Histone deacetylase inhibitor downregulation of Bcl-XL gene expression leads to apoptotic cell death in mesothelioma. Am. J. Respir. Cell. Mol. Biol. 25, 562–568 (2001).
Zhu, W. G., Lakshmanan, R. R., Beal, M. D. & Otterson, G. A. DNA methyltransferase inhibition enhances apoptosis induced by histone deacetylase inhibitors. Cancer Res. 61, 1327–1333 (2001).
Weiser, T. S. et al. Sequential 5-aza-2′-deoxycytidine-depsipeptide FR901228 treatment induces apoptosis preferentially in cancer cells and facilitates their recognition by cytolytic T lymphocytes specific for NY–ESO1. J. Immunother. 24, 151–161 (2001).
Amin, H. M., Saeed, S. & Alkan, S. Histone deacetylase inhibitors induce caspase-dependent apoptosis and downregulation of DAXX in acute promyelocytic leukaemia with t(15;17). Br. J. Haematol. 115, 287–297 (2001)
Trapani, J. A. et al. Perforin-dependent nuclear entry of granzyme B precedes apoptosis, and is not a consequence of nuclear membrane dysfunction. Cell Death Differ. 5, 488–496 (1998).
Maeda, T., Towatari, M., Kosugi, H. & Saito, H. Up-regulation of costimulatory/adhesion molecules by histone deacetylase inhibitors in acute myeloid leukemia cells. Blood 96, 3847–3856 (2000).
Magner, W. J. et al. Activation of MHC class I, II, and CD40 gene expression by histone deacetylase inhibitors. J. Immunol. 165, 7017–7024 (2000).
Mishra, N., Brown, D. R., Olorenshaw, I. M. & Kammer, G. M. Trichostatin A reverses skewed expression of CD154, interleukin-10, and interferon-γ gene and protein expression in lupus T cells. Proc. Natl Acad. Sci. USA 98, 2628–2633 (2001).
Shestakova. E., Bandu, M. T., Doly, J. & Bonnefoy, E. Inhibition of histone deacetylation induces constitutive derepression of the beta interferon promoter and confers antiviral activity. J. Virol. 75, 3444–3452 (2001).
Kim, M. S. et al. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nature Med. 7, 437–443 (2001).This study shows that the antitumour activity of HDAC inhibitors is mediated by the inhibition of angiogenesis.
Sternson, S. M., Wong, J. C., Grozinger, C. M. & Schreiber, S. L. Synthesis of 7,200 small molecules based on a substructural analysis of the histone deacetylase inhibitors trichostatin and trapoxin. Org. Lett. 3, 4239–4242 (2001).
Meinke, P. T. & Liberator, P. Histone deacetylase: a target for antiproliferative and antiprotozoal agents. Curr. Med. Chem. 8, 211–235 (2001).
Jung, M. Inhibitors of histone deacetylase as new anticancer agents. Curr. Med. Chem. 8, 1505–1511 (2001).
Remiszewski, S. W. et al. Inhibitors of human histone deacetylase: synthesis and enzyme and cellular activity of straight chain hydroxamates. J. Med. Chem. 45, 753–757 (2002).
Richon, V. M. et al. A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases. Proc. Natl Acad. Sci. USA 95, 3003–3007 (1998).The characterization of the production and function of a new class of HDAC inhibitor.
Butler, L. M. et al. Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo. Cancer Res. 60, 5165–5170 (2000).
Reed, J. C. Dysregulation of apoptosis in cancer. J. Clin. Oncol. 17, 2941–2953 (1999).
Takakura, M. et al. Telomerase activation by histone deacetylase inhibitor in normal cells. Nucleic Acids Res. 29, 3006–3011 (2001).
Cress, W. D. & Seto, E. Histone deacetylases, transcriptional control, and cancer. J. Cell. Physiol. 184, 1–16 (2000).
Terao, Y. et al. Sodium butyrate induces growth arrest and senescence-like phenotypes in gynecologic cancer cells. Int. J. Cancer 94, 257–267 (2001).
Vigushin, D. M. et al. Trichostatin A is a histone deacetylase inhibitor with potent antitumor activity against breast cancer in vivo. Clin. Cancer Res. 7, 971–976 (2001).
Coffey, D. C. et al. The histone deacetylase inhibitor, CBHA, inhibits growth of human neuroblastoma xenografts in vivo, alone and synergistically with all-trans retinoic acid. Cancer Res. 61, 3591–3594 (2001).
Ueda, H. et al. FR901228, a novel antitumor bicyclic depsipeptide produced by Chromobacterium violaceum No. 968.III. Antitumor activities on experimental tumors in mice. J. Antibiot. (Tokyo) 47, 315–323 (1994).
Saito, A. et al. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl Acad. Sci. USA 96, 4592–4597 (1999).
Kosugi, H. et al. In vivo effects of a histone deacetylase inhibitor, FK228, on human acute promyelocytic leukemia in NOD/SHI–SCID/SCID mice. Jpn J. Cancer Res. 92, 529–536 (2001).
Gilbert, J. et al. A Phase I dose escalation and bioavailability study of oral sodium phenylbutyrate in patients with refractory solid tumor malignancies. Clin. Cancer Res. 7, 2292–2300 (2001).
Carducci, M. A. et al. A Phase I clinical and pharmacological evaluation of sodium phenylbutyrate on an 120-hr infusion schedule. Clin. Cancer Res. 7, 3047–3055 (2001).
Warrell, R. P. Jr, He, L. Z., Richon, V., Calleja, E. & Pandolfi, P. P. Therapeutic targeting of transcription in acute promyelocytic leukemia by use of an inhibitor of histone deacetylase. J. Natl Cancer Inst. 90, 1621–1625 (1998).This paper shows the use of HDAC inhibitors for the treatment of APL.
Piekarz, R. L. et al. Inhibitor of histone deacetylation, depsipeptide (FR901228), in the treatment of peripheral and cutaneous T-cell lymphoma: a case report. Blood 98, 2865–2868 (2001).
Ruefli, A. A. Suberoylanilide hydroxamic acid (SAHA) overcomes multidrug resistance and induces cell death in P-gp expressing cells. Int. J. Cancer. (in the press).
Greenberg, V. L., Williams, J. M., Cogswell, J. P., Mendenhall, M. & Zimmer, S. G. Histone deacetylase inhibitors promote apoptosis and differential cell cycle arrest in anaplastic thyroid cancer cells. Thyroid 11, 315–325 (2001).
Harbour, J. W. & Dean, D. C. Chromatin remodeling and Rb activity. Curr. Opin. Cell Biol. 12, 685–689 (2000).An excellent review, which outlines the importance of HDACs for transcriptional repression by RB.
Puri, P. L. et al. Class I histone deacetylases sequentially interact with MyoD and pRb during skeletal myogenesis. Mol. Cell 8, 885–897 (2001).
Kihara-Negishi, F. et al. In vivo complex formation of PU.1 with HDAC1 associated with PU.1-mediated transcriptional repression. Oncogene 20, 6039–6047 (2001).
Ashburner, B. P., Westerheide, S. D. & Baldwin, A. S. Jr. The p65 (RelA) subunit of NF-κB interacts with the histone deacetylase (HDAC) corepressors HDAC1 and HDAC2 to negatively regulate gene expression. Mol. Cell. Biol. 21, 7065–7077 (2001).
Fuks, F., Burgers, W. A., Godin, N., Kasai, M. & Kouzarides, T. DNMT3A binds deacetylases and is recruited by a sequence-specific repressor to silence transcription. EMBO J. 20, 2536–2544 (2001).
Chen, L., Fischle, W., Verdin, E. & Greene, W. C. Duration of nuclear NF-κB action regulated by reversible acetylation. Science 293, 1653–1657 (2001).
Rountree, M. R., Bachman, K. E. & Baylin, S. B. DNMT1 binds HDAC2 and a new co-repressor, DMAP1, to form a complex at replication foci. Nature Genet. 25, 269–277 (2000).
Yarden, R. I. & Brody, L. C. BRCA1 interacts with components of the histone deacetylase complex. Proc. Natl Acad. Sci. USA 96, 4983–4988 (1999).
Doetzlhofer, A. et al. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol. Cell. Biol. 19, 5504–5511 (1999).
Ng, H. H. et al. MBD2 is a transcriptional repressor belonging to the MeCP1 histone deacetylase complex. Nature Genet. 23, 58–61 (1999).
Coull, J. J. et al. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 74, 6790–6799 (2000).
Robertson, K. D. et al. DNMT1 forms a complex with RB, E2F1 and HDAC1 and represses transcription from E2F-responsive promoters. Nature Genet. 25, 338–342 (2000).
Luo, J., Su, F., Chen, D., Shiloh, A. & Gu, W. Deacetylation of p53 modulates its effect on cell growth and apoptosis. Nature 408, 377–381 (2000).An outline of the regulation of p53 function by factor acetyltransferase activity.
Kim, G. D. et al. Sensing of ionizing radiation-induced DNA damage by ATM through interaction with histone deacetylase. J. Biol. Chem. 274, 31127–31130 (1999).
Acknowledgements
I apologise to those whose work was not cited or discussed due to space limitations. I thank S. Russell, A. Ruefli and members of my laboratory for helpful discussions, and E. Baker for help with Figure 1. R.W.J. is a Wellcome Trust Senior Research Fellow and is supported by the National Health and Medical Research Council of Australia.
Author information
Authors and Affiliations
Related links
Related links
DATABASES
Cancer.gov
LocusLink
Medscape DrugInfo
OMIM
<i>Saccharomyces</i> Genome Database
FURTHER INFORMATION
Boston University School of Medicine
Glossary
- APOPTOSIS
-
Also termed 'programmed cell death', is a natural physiological process that occurs during disease and development, and is initiated by cells in response to environmental stresses. It is characterized morphologically by membrane blebbing, chromatin condensation, loss of cell volume and DNA fragmentation, and biochemically by caspase activation.
- ONCOGENE
-
A normal gene that stimulates appropriate cell growth under normal conditions. When mutated or overexpressed, it can induce the uncontrolled proliferation of cells in the absence of growth signals and mediate neoplastic transformation.
- TUMOUR-SUPPRESSOR GENE
-
A gene that inhibits cell-cycle progression or induces apoptosis to regulate cell numbers. Often mutated or functionally inactivated in cancer.
- EPIGENETIC
-
A change that influences phenotype without altering genotype.
- DIFFERENTIATION
-
The structural and functional specialization of cells and tissues during development. It occurs by the gradual maturation of cells with specialized structures and functions from unspecialized precursors as a result of changes in gene expression.
- CELL CYCLE
-
The sequence of stages — mitosis (M), gap 1 (G1), the DNA-synthesis stage (S) and gap 2 (G2) — that an actively growing cell passes through between the time it is formed and the time it divides to give two new cells. During this time, it doubles its cytoplasmic constituents, replicates its DNA and finally divides to give two daughter cells.
- ANGIOGENESIS
-
The growth of new blood vessels from pre-existing ones. A complex phenomenon that is required for the continued growth and survival of solid neoplasms.
- CHROMATIN REMODELLING
-
An alteration in chromatin structure that affects the nuclease sensitivity of a region of chromatin. Accomplished by covalent modification of histones and/or the action of ATP-dependent remodelling complexes.
- CHEMOTHERAPY
-
Initially defined by Paul Ehrlich as “the use of a drug to combat an invading parasite without damaging the host”. Now commonly refers to the treatment of cancer using drugs that are selectively toxic for the cancerous cells.
- CHROMOSOMAL TRANSLOCATION
-
A genetic rearrangement in which part of a chromosome is detached and transferred to another chromosome, or to another portion of the same chromosome. Reciprocal translocation is when two chromosomes exchange DNA.
- LEUKAEMIA
-
A chronic or acute haematopoietic cancer that is characterized by unrestrained growth and loss of differentiation of leukocytes and their precursors. Leukaemia is classified according to the dominant cell type and severity of the disease.
- ZINC FINGER
-
A protein domain that contains two invariant cysteine, and two invariant histidine, residues that bind a single zinc atom. These domains often confer the DNA-binding component of transcription factors, but can mediate protein–protein interactions.
- PHARMACOKINETICS
-
The study of the metabolism and action of drugs, with particular emphasis on the time that is required for absorption, duration of action, distribution in the body and method of excretion.
- BIOAVAILABILITY
-
The rate and extent to which an active drug enters the general circulation, thereby permitting access to the site of action. Determined by measuring the concentration of the drug in body fluids, or by the magnitude of the pharmacological response.
- DIFFERENTIAL DISPLAY
-
A method for identifying differentially expressed genes using anchored oligo-dT and random oligonucleotide primers and polymerase chain reaction (PCR) on reverse-transcribed RNA from different cell populations. The amplified complementary DNAs are displayed, and comparisons are drawn between the different cell populations.
- DNA MICROARRAY
-
A high-throughput, differential gene-expression screen of complementary DNA or oligonucleotide libraries that are printed in extremely high density on microchips. These microchips are probed with a mixture of fluorescently tagged cDNAs that are produced from two different cell populations, and analysed with a laser confocal scanner.
- CHECKPOINT
-
A point at which the cell cycle can be halted until conditions are suitable for the cell to proceed to the next stage.
- KINETOCHORE
-
A structural feature of the chromosome to which microtubules of the mitotic spindle attach.
- CASPASES
-
A family of cysteine proteases that cleave various cellular substrates, which leads to the morphological changes that are associated with apoptosis. Might also activate inflammatory cytokines such as interleukin-1.
- TELOMERASE
-
An RNA-containing enzyme complex that extends chromosome ends (telomeres) by copying its RNA sequence repeatedly into chromosomal DNA.
Rights and permissions
About this article
Cite this article
Johnstone, R. Histone-deacetylase inhibitors: novel drugs for the treatment of cancer. Nat Rev Drug Discov 1, 287–299 (2002). https://doi.org/10.1038/nrd772
Issue Date:
DOI: https://doi.org/10.1038/nrd772
This article is cited by
-
Targeting HDAC3 to overcome the resistance to ATRA or arsenic in acute promyelocytic leukemia through ubiquitination and degradation of PML-RARα
Cell Death & Differentiation (2023)
-
Pharmacophore-based virtual screening of ZINC database, molecular modeling and designing new derivatives as potential HDAC6 inhibitors
Molecular Diversity (2023)
-
Chromatin alterations during the epididymal maturation of mouse sperm refine the paternally inherited epigenome
Epigenetics & Chromatin (2022)
-
Bioinformatics analysis of prognostic value and immunological role of MeCP2 in pan-cancer
Scientific Reports (2022)
-
Oroxylin A inhibits the migration of hepatocellular carcinoma cells by inducing NAG-1 expression
Acta Pharmacologica Sinica (2022)
