
Abstract
The prefrontal cortex (PFC) is believed to play an important role in the cognitive impairments observed in schizophrenia and has also been shown to be involved in the modulation of prepulse inhibition (PPI), a measure of preattentive information processing that is impaired in schizophrenic individuals. Phencyclidine (PCP), a noncompetitive inhibitor of the NMDA receptor, exerts psychotomimetic effects in humans, disrupts PPI, and causes hypofrontality in rodents and monkeys. We have previously demonstrated that interfering with the production of nitric oxide (NO) can prevent a wide range of PCP-induced behavioral deficits, including PPI disruption. In the present study, the role of NO signaling for the behavioral and biochemical effects of PCP was further investigated. Dialysate from the medial PFC of mice receiving systemic treatment with PCP and/or the NO synthase inhibitor, NG-nitro-L-arginine methyl ester (L-NAME, 40 mg/kg), was analyzed for cGMP content. Furthermore, a specific inhibitor of NO-sensitive soluble guanylyl cyclase (sGC), 1H-(1,2,4)oxadiazolo(4,3-a)quinoxalin-1-one (ODQ, 0.01–1 mM), was administered into the medial PFC of mice in combination with systemic injections of PCP, followed by PPI and locomotor activity testing. PCP (5 mg/kg) caused an increase in prefrontal cGMP that could be attenuated by pretreatment with the NO synthase inhibitor, L-NAME. Moreover, bilateral microinjection of the sGC inhibitor, ODQ, into the medial PFC of mice attenuated the disruption of PPI, but not the hyperlocomotion, caused by PCP. The present study shows that NO/sGC/cGMP signaling pathway in the medial PFC is involved in specific behavioral effects of PCP that may have relevance for the disabling cognitive dysfunction found in patients with schizophrenia.
Similar content being viewed by others

INTRODUCTION
Schizophrenia-spectrum patients commonly display deficits in sensorimotor gating reflecting a disturbed perception of sensory information. This has led to the hypothesis that these patients are afflicted by impairment in information processing mechanisms resulting in sensory flooding followed by a cognitive fragmentation (McGhie and Chapman, 1961; Braff et al, 1978; Freedman et al, 1987; Braff, 1993). Sensorimotor gating deficits, as measured by prepulse inhibition (PPI), and dysfunctions in cognitive domains such as social cognition and reasoning appear to be closely related in some, but not all, clinical studies of schizophrenia (Perry et al, 1999; Wynn et al, 2005). Phencyclidine (PCP), a noncompetitive NMDA receptor antagonist, can induce a state in humans that closely resembles schizophrenia, encompassing both positive and negative symptoms as well as cognitive dysfunctions (Luby et al, 1959; Yesavage and Freman, 1978; Javitt and Zukin, 1991). Administration of PCP to laboratory animals is commonly used to model certain aspects of schizophrenia, including the sensorimotor gating deficits found in this disease (Mansbach and Geyer, 1989; Klamer et al, 2001; Linn et al, 2003; Fejgin et al, 2007). Thus, studies of the mechanisms by which PCP disrupts PPI may have heuristic value when trying to identify the underlying mechanisms behind sensorimotor gating deficits in schizophrenic patients. In addition, subchronic PCP administration has been shown to induce hypofrontality in rodents and monkeys, which is thought to bear relevance for the cognitive dysfunction observed in schizophrenia (Jentsch et al, 1997a, 1997b).
The neuroanatomical substrates that mediate the disruption of PPI by PCP and similar agents have not yet been fully characterized, although both subcortical structures and prefrontal regions appear to be involved (Bakshi and Geyer, 1998). The prefrontal cortex (PFC) has been extensively linked to several cognitive domains such as executive function and working memory, and a disturbance of this region and its circuitry is considered a core feature of the cognitive dysfunction observed in schizophrenia (Elvevag and Goldberg, 2000). The PFC constitutes a heterogeneous region and can thus be separated into functionally diverse subregions. It is extensively interconnected with the rest of the brain and is commonly divided into a medial, lateral, and ventral part in rodents (Dalley et al, 2004). The medial PFC is thought to have a modulatory effect on sensorimotor gating in rodents as selective dopaminergic lesions in this area produce deficits in PPI in rats (Bubser and Koch, 1994). In addition, neurotoxic lesions in the medial PFC of rats have been shown to prevent the PPI-disruptive effects of the NMDA receptor antagonist, dizocilpine (MK-801), whereas the disruptive effect of the dopamine receptor agonist apomorphine was not affected by such lesions (Schwabe and Koch, 2004). This indicates that the medial PFC may be an important structure implicated in the effects of NMDA receptor antagonists on preattentive information processing.
Nitric oxide (NO) is a gaseous messenger molecule that can serve both as an extracellular and intracellular transmitter in the brain, most likely through modulation of classical neurotransmitter release (Kano et al, 1998; Segovia and Mora, 1998; Smith and Whitton, 2000, 2001; Wegener et al, 2000; Prast and Philippu, 2001). The main effector of NO signaling is cGMP formed by soluble guanylyl cyclase (sGC) (Karatinos et al, 1995) even if NO, to a lesser extent, may act through cAMP formation by adenylyl cyclase and protein nitrosylation. Furthermore, an early in vitro study by Vesely (1979) shows that PCP readily activates guanylyl cyclase in rat brain tissue. We have previously demonstrated that a wide range of PCP-induced behavioral deficits, including PPI, habituation of acoustic startle, latent inhibition, spatial learning, spatial reference memory, and working memory, all can be prevented by interfering with the production of NO (Johansson et al, 1997, 1998; Klamer et al, 2001, 2004b, 2004a, 2004c, 2005a; Wass et al, 2006a, 2006b; Palsson et al, 2007). This suggests that the disruptive effect of PCP on preattentive information processing and cognitive function in rodents is, in part, mediated by an increase in NO activity. Hence, neuronal nitric oxide synthase (nNOS) may prove a valuable pharmacological target when developing novel treatment options for the disabling cognitive dysfunctions found in schizophrenia. The possibility that a dysfunctional NO system is involved in this disease is strengthened by studies showing an abnormal distribution of nitrinergic neurons in the frontal and temporal lobes of schizophrenic patients, which may reflect that the normal pattern of neuronal migration during development of the cerebral cortex may be affected in these patients (Akbarian et al, 1993a, 1993b). More recent studies show that polymorphisms in the nNOS gene are associated with schizophrenia and PFC function in schizophrenic patients (Shinkai et al, 2002; Reif et al, 2006). In addition, two recent studies show a significant increase of NO metabolites in the serum of schizophrenic patients indicating that NO function may be dysregulated in this disorder (Taneli et al, 2004; Yilmaz et al, 2007) even if the interpretation of these findings is complicated by earlier studies showing a decrease in NO activity (Srivastava et al, 2001; Suzuki et al, 2003).
In the present study, the role of NO signaling for the behavioral and biochemical effects of PCP was further investigated. To obtain a biochemical estimate of local NO signaling in the medial PFC, dialysate from the medial PFC of mice receiving systemic treatment with PCP and/or the NOS-inhibitor, NG-nitro-L-arginine methyl ester (L-NAME), was analyzed for cGMP content. Furthermore, the specific inhibitor of NO-sensitive sGC, 1H-(1,2,4)oxadiazolo(4,3-a)quinoxalin-1-one (ODQ), was administered into the medial PFC of a second group of mice in combination with systemic injections of PCP and thereafter tested for PPI and locomotor activity.
MATERIALS AND METHODS
Animals
Male NMRI mice (Charles River, Germany), 25–35 g, were used. The animals were housed, six per cage, in a colony room under constant temperature (20±1°C) and humidity (50±5%). Food and water were available ad libitum. The daylight cycle was maintained artificially (lights on from 0600 to 1800 hours), and the behavioral experiments were conducted during the light phase. All experimental procedures used in the present study were approved by the Ethics Committee for Animal Experiments, Göteborg, Sweden.
Drugs
Phencyclidine hydrochloride (PCP, Sigma Chemicals, St Louis, MO, USA), ketoprofen (Veter, Sweden), L-NAME (RBI, Natick, USA), and ODQ (Sigma-Aldrich, Stockholm, Sweden) were used in the present study. PCP and L-NAME were dissolved in saline (0.9% NaCl) and injected intraperitoneally (i.p.). ODQ was dissolved in 100% DMSO and stored in batches at −20°C and diluted in Ringer's solution on the day of testing, reaching a final DMSO content of 1%.
Probes
All probes were manufactured on site. A modified version of the microdialysis probes described by Santiago and Westerink (1990) was used. The inlet and the outlet of the probe were made of PE20 polyethylene tubing with an o.d./i.d. of 1.09/0.38 mm (Becton Dickinson and Company, Sparks, MD, USA). From the inlet a fused silica was extended approximately 3 mm. Centered between the inlet and outlet a glass rod was attached with Super Epoxy (Loctite, Göteborg, Sweden).
To facilitate the probe production procedure, special stands (Medi Tech, Göteborg University, Sweden) were used. The dialysis membrane, prepared from a copolymer of polyacrylonitrile and sodium methallyl sulfonate with an o.d./i.d. of 310/220 μm (Hospal industrie, Meyzieu, France), was thread on the fused silica inlet, and sealed with a plug of glue. The length of the exchange area was 1.5 mm. The remaining membrane area was covered with silicon glue (CAF 3, Rhodorsil Silicones, Saint-Fons Cedex, France). The dialysis probes were thereafter connected to a microperfusion pump (Univentor 864 Syringe Pump, Univentor, Zejtan, Malta) and perfused at a flow rate of 2 μl/min, with 70% ethanol for 20 min and subsequently with Ringer's solution for 60 min. Finally the inlet and the outlet of the dialysis probe were sealed by heating. This procedure was done to wash and prepare the probes for implantation. The dialysis probes were stored in Ringer's solution for a maximum of 3 days at 6°C before implantation.
Surgical Procedure
The mice were anesthetized with isoflurane (Isofluran Baxter; Apoteket AB, Sweden), placed in a Kopf stereotaxic instrument (David Kopf Instruments, Tujunga, CA, USA), and kept on a heating pad to prevent hypothermia. For local infusion experiments the skull was exposed, and two holes for the guide cannulas (stainless steel, length 10 mm, with an o.d./i.d. of 0.6/0.45 mm), and one hole for an anchor screw were drilled. The coordinates used for the medial PFC region relative to the bregma were as follows: anterior+1.8 mm, lateral to midline±0.8 mm, and ventral −1.0 mm from the brain surface (Franklin and Paxinos, 1996). The dura was removed using a sharp needle, and the guide cannulas and the anchor screw were secured with dental cement (Dentalon plus, AgnTho's AB, Lidingö, Sweden). For the microdialysis experiments the skull was similarly exposed and two holes, one for the dialysis probe (either right or left hemisphere) and one for the anchor screw, were drilled. The anterior and lateral coordinates for the medial PFC region relative to the bregma were identical to above and the ventral coordinate was −1.6 mm from the brain surface (Franklin and Paxinos, 1996). As above the dura was removed by using a sharp needle, and the probe and the anchor screw were secured with dental cement. After surgery, the mice were administered 1.0 ml of saline, subcutaneously (s.c.), to avoid postoperative dehydration. A total of 10 mg/kg/ml of ketoprofen was administered s.c. as a prophylactic analgesic. The mice were then allowed to recover for 3–4 (local infusion) or 2 (microdialysis) days before the experiment. They were housed individually in standard plastic cages (Macrolon III; 400 × 250 × 150 mm). Surgery started 1 week after the arrival of the animals from the breeder.
Microdialysis Experiments
The animals (n=40) were divided into four treatment groups: sal+sal (n=10); sal+PCP (n=10); L-NAME+sal (n=10); and L-NAME+PCP (n=10). They were connected to the microdialysis apparatus via a liquid swivel (FEB-tubing, CMA/Microdialysis AB, Stockholm, Sweden) and were able to move freely during the experiment. The dialysis probes were perfused with Ringer's solution, at a constant rate of 1.5 μl/min, for a 60 min habituation period to establish a stable baseline. Thereafter, dialysate samples (135 μl) were collected over two 90 min periods (Vial Plastic 300 ml, CMA/Microdialysis AB. Saline (10 ml/kg) or L-NAME (40 mg/10 ml/kg) was administered 80 min after the start of the first of these sampling periods, followed by saline (10 ml/kg) or PCP (5 mg/10 ml/kg) 10 min later. The dialysate samples were stored at −35°C until assayed for cGMP using an enzyme immunoassay. Five animals were excluded due to technical problems during the microdialysis experiments.
Immunoassay
The cGMP amount was assessed using a commercial enzyme immunoassay kit (CG-201 cGMP Enzyme Immunoassay Kit, Sigma-Aldrich). The acetylated version of the protocol of the manufacturer was followed with a single modification. The standard range was shifted from 100–0.16 to 20–0.032 pmol. The assay is based on the competition between sample cGMP and a fixed quantity of cGMP, with an alkaline phosphatase molecule covalently bound to it, for a limited number of binding sites on a cGMP-specific antibody. A fixed amount of substrate is then added and after a short incubation period the enzyme reaction is stopped and the intensity of the resulting yellow color is read on a microplate reader at 405 nm. The intensity of the bound color is inversely proportional to the amount of cGMP in either standards or samples. All standards and samples were analyzed in duplicates and the mean readings were used to calculate cGMP content. Four animals were excluded due to technical problems with the cGMP immunoassay.
Data and statistical analysis
The consecutive cGMP contents of the dialysis samples were used as data. The cGMP content of the second sample (90–180 min) was divided by the content of the first sample (0–90 min) and multiplied by 100 to obtain the change in cGMP level relative to the baseline. To investigate whether the drug treatments had any significant effect on cGMP levels, a two-way analysis of variance (ANOVA) was performed followed by Bonferroni's post hoc test to specifically assess the difference between the different treatment groups. Two-tailed levels of significance were used and p<0.05 was considered statistically significant. Two animals, treated with sal-PCP, were excluded from the data analysis as they were labeled as outliers by a box-plot-based outlier test (>1.5 box lengths from the box edge).
Probe placement verification
After termination of the experiments the rats were decapitated. The brains were removed and fixated (Accustain, Sigma-Aldrich) for approximately 1 week. Probe placement was verified by sectioning the brains using a vibratome and an atlas of the mouse brain for reference (Franklin and Paxinos, 1996). Only animals with correct probe placement in the medial PFC were included in the analysis (Figure 1a).

Coronal mouse brain section of the medial PFC (+2.1 mm from bregma) showing a schematic drawing of (a) estimated probe placement for the microdialysis experiments. The hatched area represents the semipermeable part of the membrane (b) estimated guide/cannula placement. Reprinted from The Mouse Brain in Stereotaxic Coordinates, KBJ Franklin and G Paxinos, Figure 18, Copyright (1996), with permission from Elsevier.
Prepulse Inhibition Experiments
Apparatus
Acoustic startle was recorded by an MOPS 2b startle response recording system (Metod och Produkt, Svenska AB, Göteborg, Sweden). The animals were placed in small wire-mesh cages (10 × 5.5 × 5.5 cm) made of stainless steel, which were suspended at one point at the top to a piston in such a way that they could move freely under the piston. A sudden movement of the animal inside the cage caused a displacement of the piston, the acceleration of which was converted to an analog signal by a moving coil transducer. This signal was sampled and digitized with a 12-bit analog-to-digital (A/D) resolution by a microcomputer, which also served to control the delivery of acoustic stimuli. Startle amplitude was defined as the maximum signal amplitude (A/D units) that occurred during the first 40 ms after delivery of the startle-eliciting stimulus. Three cages were used simultaneously and each cage was housed in a separate, dimly lit, and sound-attenuated cabinet (52 × 42 × 38 cm). The cages were calibrated for equal sensitivity before test and a mouse tested in one cage was always tested in the same cage at subsequent tests. The acoustic signal consisted of white noise delivered to the animal by two high-frequency loudspeakers built into the ceiling of the cabinet.
PPI paradigm
Each test session started with a 10-min adaptation period containing only white background noise 62 dB(A). The background noise was interrupted at stimulus presentations by a burst of white noise with a rise/decay time of less than 1 ms. Startle pulse intensity was set to 105 dB(A) and prepulse intensity to 70 dB(A). Startle pulse duration was set to 20 ms and prepulse duration to 60 ms. The prepulse was presented immediately before the startle pulse. After the 10-min adaptation period, the animals were presented with a series of five startle pulse-alone trials followed by a series of five prepulse-alone trials. The pulse-alone trials served only to accommodate the animals to the sudden change in stimulus conditions and were omitted from the data analysis and the prepulse-alone trials were analyzed only to ensure that these stimuli did not evoke any startle responses on their own. Thereafter the animals were presented, three times repeatedly, with a series of five prepulse-pulse trials followed by a series of five pulse-alone trials, ie a total of 30 trials. The time between trials was always 10 s and the time between any series of trials was 70 s.
Testing procedure
The mice were pretested without drug treatment before surgery to ensure that they displayed basal startle reactivity. At the day of the experiment a dummy cannula was carefully inserted and retracted into the guide cannulas to reduce the risk for spreading depression. This procedure was used because previous observations in our laboratory indicate that cannula insertion is commonly accompanied by spreading depression, and the probability of a new spreading depression is largely reduced at the second insertion. The animals (n=72) were divided into eight treatment groups: veh+sal (n=11); veh+PCP (n=14); ODQ 0.01 mM+sal (n=9); ODQ 0.1 mM+sal (n=6); ODQ 1 mM+sal (n=7); ODQ 0.01 mM+PCP (n=10); ODQ 0.1 mM+PCP (n=6); and ODQ 1 mM+PCP (n=9). The veh+sal group and the veh+PCP group were both larger since these groups had to be represented in every new experimental session. Each mouse was tested twice; a control test followed by a drug test. At the time of experiment, the dummy cannula was inserted 1.0 mm ventral to the brain surface. After this, the mice were allowed to rest for 60 min in their home cages before the first PPI test, which served as a control test. The animals were injected locally, with either 0.5 μl ODQ (1, 0.1, or 0.01 mM) or vehicle (1% DMSO) bilaterally in the medial PFC 30 min after the end of the first test. The local injection lasted 1 min and the cannula was left in place for another minute to allow diffusion of the drug. PCP (5 mg/kg) or saline was then administered (i.p.) and 5 min later the animals were placed in the startle chambers for the second PPI test.
Guide placement verification
After termination of the experiments the mice were decapitated. The brains were removed and fixated (Accustain) for approximately 1 week. Guide placement was verified by sectioning the brains using a vibratome and an atlas of the mouse brain for reference (Franklin and Paxinos, 1996). Only animals with correct cannula placement in the medial PFC were included in the analysis (Figure 1b).
Data analysis
The mean response amplitude for pulse-alone trials (P) was calculated for each mouse and test. This measure was used in the statistical analysis to assess drug-induced changes in acoustic startle response (ASR). The mean response amplitude for prepulse-pulse trials (PP) was also calculated and used to express the percent PPI according to the following formula:

using this formula, a 0% value denotes no difference between pulse-alone and prepulse-pulse response amplitudes and consequently no PPI. The Drug test/Control test PPI ratios for each animal were then used to express the change in PPI compared to the control test. This procedure was used to compensate for the interindividual differences in basal PPI between the animals. The same rationale was also used for assessment of the ASR. Statistical analysis was performed using a two-way ANOVA with pretreatment (veh or ODQ) and treatment (sal or PCP) as fixed factors to assess the effects of the highest dose of ODQ (1 mM) on PCP-induced PPI deficits. When introducing Control test PPI as a covariate, it did not significantly affect the ANOVA and was thus not considered further in the statistical analysis. Bonferroni's post hoc tests were conducted to specifically assess the difference between treatment groups for all doses. Two-tailed levels of significance were used and p<0.05 was considered statistically significant. Two animals (one mouse treated with sal+ODQ 0.01 mM and one mouse treated with sal+ODQ 1 mM) were excluded from the data analysis as they were labeled as outliers by a box-plot-based outlier test (>1.5 box lengths from the box edge).
Locomotor Experiments
Apparatus
Locomotor activity of the mice was recorded using eight box-shaped Plexiglas devices with a floor area of 42 × 42 cm. The activity boxes were housed in dimly lit and sound-attenuated cabinets (420 × 420 × 45 cm). A computer-based system determined the horizontal location of the animal at all times using five times five rows of photocell beams.
Testing procedure
The animals (n=30) were divided into four treatment groups: veh+sal (n=6); veh+PCP (n=8); ODQ 0.1 mM+sal (n=8); and ODQ 0.1 mM+PCP (n=8). After the insertion of a dummy cannula (as described above) to control for spreading depression, the mice were placed in the activity boxes and allowed to habituate for 60 min before injection. The animals were then administered either ODQ (0.1 mM) or vehicle in analogy with the PPI experiments and placed in the activity boxes for 10 min before an i.p. injection with either PCP (5 mg/kg) or saline. After this, the mice were placed in the activity boxes and their locomotion, defined as the accumulated number of crossed photocell beams, was recorded for 60 min. The first 30 min of this recording period was excluded from the analysis, to reduce the influence of injection-induced hypermotility and to allow PCP to exert its effect. Verification of cannula placement was performed as described above for the PPI experiments.
Data analysis
Two-way repeated-measures ANOVA was performed to investigate whether pretreatment with ODQ interacted with the effects of PCP on locomotor activity. The analysis used two-tailed levels of significance and p<0.05 was considered statistically significant.
RESULTS
Microdialysis Experiments
The effect of systemic PCP treatment on cGMP levels in the medial PFC
Prefrontal cGMP levels were significantly increased by an average of 56% when comparing PCP (5 mg/kg, i.p.)-treated animals to control animals (p=0.034, Bonferroni's test) (Figure 2).

Pretreatment with NG-nitro-L-arginine methyl ester (L-NAME) prevents the phencyclidine (PCP)-induced increase in cGMP levels. Effects of L-NAME and/or PCP on cGMP levels in the medial PFC. Data expressed as mean values relative to preceding baseline +/− SEM (*p<0.05, Bonferroni's test).
The effect of pretreatment with L-NAME on the PCP-induced increase in PFC cGMP levels
L-NAME (40 mg/kg, i.p.) did not cause any significant changes in prefrontal cGMP levels by itself (p=0.959, Bonferroni's test) (Figure 2). Furthermore, the expected increase in cGMP caused by PCP was antagonized by pretreatment with L-NAME, as evidenced by a significant pretreatment × treatment interaction effect (two-way ANOVA, F(1, 27)=5.251, p=0.030). This interpretation was further supported by the observation that animals treated with L-NAME+PCP displayed significantly lower cGMP levels than animals treated with sal+PCP (p=0.038, Bonferroni's test) but did not differ from control animals (p=0.999, Bonferroni's test).
Prepulse Inhibition Experiments
The effect of local pretreatment with ODQ in the PFC on PCP-induced disruption of PPI
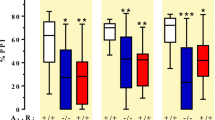
Statistical analysis showed a significant effect of PCP (5 mg/kg, i.p.) treatment in the data set (two-way ANOVA, effect of treatment, F(1, 37)=5.359, p=0.026) (Figure 3). Furthermore, a significant pretreatment × treatment interaction effect could be demonstrated for the highest dose of ODQ (1 mM) suggesting that ODQ at this dose blocked the effects of PCP. This motivated further investigation of the data set using post hoc comparisons to assess difference between treatment groups. Mice subjected to PCP showed a decrease in their PPI response compared to saline and thus a significant decrease in their Drug/Control ratio (p=0.002, Bonferroni's test) compared to saline treatment. This effect was not prevented by pretreatment with the lower dose of ODQ (0.01 mM) (p=1.000, Bonferroni's test). Pretreatment with a higher dose of ODQ (0.1 mM) blocked the effect of PCP since this group differed significantly from PCP-treated animals (p=0.036, Bonferroni's test) but not from control animals. An additional dose of ODQ (1 mM) was also tested and blocked the PCP-induced deficit in analogy with the 0.1 mM dose (p=0.038, Bonferroni's test).

Local pretreatment with 1H-(1,2,4)oxadiazolo(4,3-a)quinoxalin-1-one (ODQ) blocks phencyclidine (PCP)-induced disruption of prepulse inhibition (PPI). Effects of bilateral, prefrontal ODQ treatment followed by systemic PCP treatment on PPI. Pretreatment with ODQ in the dose range 0.01–1 mM did not significantly affect PPI by itself (data not shown for 0.01–0.1 mM). Data represented as mean PPI ratios (Drug test/Control test) +/− SEM for each treatment group (**p<0.01, *p<0.05, Bonferroni's test).
The effect of local ODQ treatment in the PFC on PPI
The highest dose of ODQ (1 mM) did not have any significant effect on PPI ratios in the present study (two-way ANOVA, effect of pretreatment, F(1, 37)=2.611, p=0.115) and the post hoc test did not reveal any change in PPI ratio, compared to the control group (p=1.000, Bonferroni's test) (Figure 3).
The effects of drug treatment on ASR reactivity
None of the drugs administered locally or systemically caused any significant changes in ASR as measured by a Drug/Control ratio (two-way ANOVA, effect of pretreatment, F(1, 40)=0.116, p=0.735; effect of treatment, F(1, 40)=0.527, p=0.527; interaction effect, F(1, 40)=1.337, p=0.254) (Figure 4).

Drug treatment does not affect acoustic startle response (ASR). Effects of bilateral, 1H-(1,2,4)oxadiazolo(4,3-a)quinoxalin-1-one (ODQ) treatment in the medial PFC followed by systemic phencyclidine (PCP) treatment on ASR. Pretreatment with ODQ in the dose range 0.01–1 mM did not significantly affect ASR by itself (data not shown for 0.01–0.1 mM). Data represented as mean ASR ratios (Drug test/Control test) +/− SEM for each treatment group. No statistically significant effects of treatment were obtained.
Locomotor Experiments
The effect of PCP treatment on locomotor activity
Statistical analysis of locomotor activity after PCP (5 mg/kg, i.p.) injection showed a significant increase in locomotor activity during the 30 min recording period (two-way ANOVA, effect of treatment, F(1, 26)=12.270, p=0.002) (Figure 5).

Local pretreatment with 1H-(1,2,4)oxadiazolo(4,3-a)quinoxalin-1-one (ODQ) does not block phencyclidine (PCP)-induced hyperlocomotion. Effects of bilateral ODQ treatment (0.1 mM) in the medial PFC on PCP-induced locomotor activity (LMA) stimulation in mice. Data represented as mean counts +/− SEM during a 30 min recording period (*p<0.05, Bonferroni's test).
The effect of local pretreatment with ODQ in the PFC on PCP-induced hyperlocomotion
Administration of ODQ (0.1 mM) into the medial PFC did not exert any significant effect by itself when compared to control animals (two-way ANOVA, effect of pretreatment, F(1, 26)=0.041, p=0.840) (Figure 5). Furthermore, pretreatment with ODQ (0.1 mM) did not prevent the hyperlocomotion caused by PCP as evidenced by the lack of a significant interaction effect (two-way ANOVA, pretreatment × treatment interaction, F(1, 26)=0.104, p=0.750).
DISCUSSION
The main findings of the present study demonstrate that the psychotomimetic drug, PCP, induces an increase in cGMP levels in the mouse medial PFC, through an NO-related mechanism. Furthermore, selective inhibition of NO-sensitive cGMP production in the mouse medial PFC markedly reduces the PCP-induced disruption of PPI, whereas the stimulatory effects of PCP on locomotor activity remain unaffected.
Effects on Prefrontal cGMP Efflux
The observed increase in prefrontal cGMP levels is probably a consequence of increased NO production, as sGC is the major target for NO signaling in the brain (Karatinos et al, 1995). Furthermore, a number of microdialysis studies have shown increases in cGMP levels in the rat frontal cortex following administration of NO donors (Laitinen et al, 1994; Pepicelli et al, 2004). Further evidence for the involvement of NO can be derived from the present study which shows that the PCP-induced increase of cGMP in the PFC can be attenuated by pretreatment with the NOS inhibitor, L-NAME. This is in line with previous studies in rats showing that prefrontal cGMP levels are lowered after L-NAME administration, in a study using the nonspecific phosphodiesterase inhibitor, isobutylmethyl xanthine, to increase cGMP levels (Laitinen et al, 1997). However, L-NAME did not affect basal cGMP levels in the present experiment. The effect of PCP on cGMP levels may be caused by an increase in prefrontal glutamate, an interpretation that is supported by the recent findings that NMDA receptor antagonists increase glutamate efflux in the PFC of rats (Moghaddam et al, 1997; Adams and Moghaddam, 1998). Thus, the present findings advocate the existence of a prefrontal NO/sGC/cGMP system that may be associated with glutamatergic neurotransmission.
Effects of Local sGC Inhibition
The disruption of PPI by PCP was prevented, in a dose-related manner, by administration of ODQ into the medial PFC. This supports the presence of a prefrontal, NO-sensitive mechanism that accounts for at least some behavioral effects of PCP. Inhibition of NO production in general appears to be able to ameliorate a wide range of PCP-induced behavioral deficits as shown in several studies by the present authors (Johansson et al, 1997; Klamer et al, 2001, 2004a, 2004c, 2005b; Wass et al, 2006a, 2006b; Palsson et al, 2007). Furthermore, the sGC inhibitor methylene blue has shown an antipsychotic profile both in animal models of NMDA hypofunction (Deutsch et al, 1996; Klamer et al, 2004b) and, more importantly, when given as an adjuvant in patients with schizophrenia (Deutsch et al, 1997). However, it is not likely that all these behavioral effects are modulated through a common and exclusively prefrontal, NO-dependent mechanism. PCP is known to exert its effects not only through the NMDA receptor, but also by, acting as an agonist on dopamine receptors and by blocking dopamine reuptake (Rothman et al, 1989; Seeman et al, 2005; Seeman and Lasaga, 2005). Nevertheless, the lack of a temporal correlation between corticolimbic dopamine levels and behavioral effects of PCP suggests that a substantial part of the effects of this compound is mediated through non-dopaminergic mechanisms (Adams and Moghaddam, 1998). A possible chain of events explaining the present results may be a prefrontal NO-mediated activation of sGC/cGMP signaling following stimulation of non-NMDA glutamate receptors caused by a PCP-induced increase in glutamatergic neurotransmission. As mentioned above, recent studies show that administration of NMDA receptor antagonists such as PCP and ketamine lead to a substantial glutamate efflux in the PFC of rats. This efflux may subsequently activate non-NMDA glutamate receptors and secondarily facilitate, eg dopaminergic neurotransmission in the PFC. Such an activation is suggested to be essential for the behavioral effects of NMDA antagonists as local application of AMPA/kainate antagonists has been shown to block the effects of PCP and ketamine on stereotypy, locomotor activity, and working memory (Moghaddam et al, 1997; Takahata and Moghaddam, 2003). In addition, preliminary studies using in vivo voltammetry and NO-sensitive microsensors (Brown and Lowry, 2003) indicate that NO production is increased in the medial PFC following systemic PCP administration in rats (Lowry JP et al in collaboration, data not published). Tentatively, an NO dysregulation in this region could affect several neurotransmitter systems and thus contribute substantially to the behavioral effects of NMDA antagonists. However, the role of the PFC for PPI appears to be of a modulatory rather than of a mediating nature, as evidenced by a relative absence of effects on PPI following exitotoxic medial PFC lesions in rats (Swerdlow et al, 1995; Schwabe and Koch, 2004; Schneider and Koch, 2005). Thus, even if a functional medial PFC is not essential for basal preattentive filter mechanisms, it may be critical for the NO-dependent effects of PCP.
Prefrontal NO Signaling and Behavioral Specificity
In the present study, local administration of ODQ in the PFC, at a dose that was effective in the PPI model, did not prevent PCP-induced hyperlocomotion. Given the strong association between subcortical dopaminergic neurotransmission and locomotion, a possible explanation may be that the prefrontal NO-sensitive neuronal circuitry discussed above does not have a major influence on the mesocorticolimbic neuronal circuitry that has been demonstrated to be strongly associated with locomotor activity (Kelly and Iversen, 1976; Clarke et al, 1988). This assumption is supported by a recent study on the behavioral effects of medial PFC lesions in rats that showed no change in locomotor activity of these animals (Schneider and Koch, 2005), suggesting that the medial PFC is not essential for basal motor activity. In summary, manipulation of nitrinergic activity in the medial PFC may primarily affect preattentive information processing functions such as PPI rather than more subcortical basal functions such as locomotion. However, since systemic pretreatment with NOS inhibitors readily prevents the hyperlocomotion caused by PCP (Johansson et al, 1997; Klamer et al, 2004a), subcortical dopaminergic activity may also be under the influence of nitrinergic neurotransmission, albeit not primarily modulated by a prefrontal circuitry.
NOS Isoforms and PCP
In addition to the above-mentioned studies that have been using the nonspecific NOS inhibitor L-NAME, other studies have reported that selective nNOS inhibitors attenuate behavioral effects of PCP in rodents (Wiley, 1998; Johansson et al, 1999; Klamer et al, 2004a). Although the selectivity of such compounds can be discussed and rather should be viewed as a strong preference for the nNOS isoform, these studies suggest an involvement of nNOS in the psychotomimetic effects of PCP. Furthermore, nNOS knockout mice appear less sensitive to the disruptive effects of PCP on PPI and PCP-induced hyperlocomotion (Wiley et al, 1999; Bird et al, 2001; Klamer et al, 2005a).
CONCLUDING REMARKS
The present findings indicate that an NO/sGC/cGMP signaling mechanism in the medial PFC is involved in specific behavioral effects of PCP that are considered relevant for the pathophysiology of cognitive dysfunction in schizophrenia. This is in line with several studies showing disturbances of NO function in the PFC of schizophrenic patients (Akbarian et al, 1993a, 1993b; Xing et al, 2002; Baba et al, 2004; Reif et al, 2006). As NO is known to have effects on the storage, uptake, and/or release of most neurotransmitters including glutamate, GABA, and dopamine (Bernstein et al, 2005), it is well positioned to play an integrative role in brain function and pathology. Furthermore, sGC, the main effector of NO, is widely distributed throughout the human and rodent brain in a complementary way to nNOS (Southam and Garthwaite, 1993; Gotti et al, 2005). A recent comparative study shows that interspecies differences in NO/sGC/cGMP signaling may be small between rodents and primates thereby implying that the NO system may serve similar functions across species (Pifarre et al, 2007). Thus, translational studies of brain NO function may prove valuable when searching for novel treatment strategies targeting sensorimotor gating deficits and cognitive dysfunctions observed in schizophrenia.
References
Adams B, Moghaddam B (1998). Corticolimbic dopamine neurotransmission is temporally dissociated from the cognitive and locomotor effects of phencyclidine. J Neurosci 18: 5545–5554.
Akbarian S, Bunney Jr WE, Potkin SG, Wigal SB, Hagman JO, Sandman CA et al (1993a). Altered distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase cells in frontal lobe of schizophrenics implies disturbances of cortical development. Arch Gen Psychiatry 50: 169–177.
Akbarian S, Vinuela A, Kim JJ, Potkin SG, Bunney Jr WE, Jones EG (1993b). Distorted distribution of nicotinamide-adenine dinucleotide phosphate-diaphorase neurons in temporal lobe of schizophrenics implies anomalous cortical development. Arch Gen Psychiatry 50: 178–187.
Baba H, Suzuki T, Arai H, Emson PC (2004). Expression of nNOS and soluble guanylate cyclase in schizophrenic brain. Neuroreport 15: 677–680.
Bakshi VP, Geyer MA (1998). Multiple limbic regions mediate the disruption of prepulse inhibition produced in rats by the noncompetitive NMDA antagonist dizocilpine. J Neurosci 18: 8394–8401.
Bernstein HG, Bogerts B, Keilhoff G (2005). The many faces of nitric oxide in schizophrenia. A review. Schizophr Res 78: 69–86.
Bird DC, Bujas-Bobanovic M, Robertson HA, Dursun SM (2001). Lack of phencyclidine-induced effects in mice with reduced neuronal nitric oxide synthase. Psychopharmacology (Berl) 155: 299–309.
Braff DL (1993). Information processing and attention dysfunctions in schizophrenia. Schizophr Bull 19: 233–259.
Braff D, Stone C, Callaway E, Geyer M, Glick I, Bali L (1978). Prestimulus effects on human startle reflex in normals and schizophrenics. Psychophysiology 15: 339–343.
Brown FO, Lowry JP (2003). Microelectrochemical sensors for in vivo brain analysis: an investigation of procedures for modifying Pt electrodes using Nafion. Analyst 128: 700–705.
Bubser M, Koch M (1994). Prepulse inhibition of the acoustic startle response of rats is reduced by 6-hydroxydopamine lesions of the medial prefrontal cortex. Psychopharmacology (Berl) 113: 487–492.
Clarke PB, Jakubovic A, Fibiger HC (1988). Anatomical analysis of the involvement of mesolimbocortical dopamine in the locomotor stimulant actions of d-amphetamine and apomorphine. Psychopharmacology (Berl) 96: 511–520.
Dalley JW, Cardinal RN, Robbins TW (2004). Prefrontal executive and cognitive functions in rodents: neural and neurochemical substrates. Neurosci Biobehav Rev 28: 771–784.
Deutsch SI, Rosse RB, Paul SM, Tomasino V, Koetzner L, Morn CB et al (1996). 7-Nitroindazole and methylene blue, inhibitors of neuronal nitric oxide synthase and NO-stimulated guanylate cyclase, block MK-801-elicited behaviors in mice. Neuropsychopharmacology 15: 37–43.
Deutsch SI, Rosse RB, Schwartz BL, Fay-McCarthy M, Rosenberg PB, Fearing K (1997). Methylene blue adjuvant therapy of schizophrenia. Clin Neuropharmacol 20: 357–363.
Elvevag B, Goldberg TE (2000). Cognitive impairment in schizophrenia is the core of the disorder. Crit Rev Neurobiol 14: 1–21.
Fejgin K, Safonov S, Palsson E, Wass C, Engel JA, Svensson L et al (2007). The atypical antipsychotic, aripiprazole, blocks phencyclidine-induced disruption of prepulse inhibition in mice. Psychopharmacology (Berl) 191: 377–385.
Franklin KBJ, Paxinos G (1996). The Mouse Brain in Stereotaxic Coordinates. Academic Press: New York.
Freedman R, Adler LE, Gerhardt GA, Waldo M, Baker N, Rose GM et al (1987). Neurobiological studies of sensory gating in schizophrenia. Schizophr Bull 13: 669–678.
Gotti S, Sica M, Viglietti-Panzica C, Panzica G (2005). Distribution of nitric oxide synthase immunoreactivity in the mouse brain. Microsc Res Tech 68: 13–35.
Javitt DC, Zukin SR (1991). Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry 148: 1301–1308.
Jentsch JD, Redmond Jr DE, Elsworth JD, Taylor JR, Youngren KD, Roth RH (1997a). Enduring cognitive deficits and cortical dopamine dysfunction in monkeys after long-term administration of phencyclidine. Science 277: 953–955.
Jentsch JD, Tran A, Le D, Youngren KD, Roth RH (1997b). Subchronic phencyclidine administration reduces mesoprefrontal dopamine utilization and impairs prefrontal cortical-dependent cognition in the rat. Neuropsychopharmacology 17: 92–99.
Johansson C, Deveney AM, Reif D, Jackson DM (1999). The neuronal selective nitric oxide inhibitor AR-R 17477, blocks some effects of phencyclidine, while having no observable behavioural effects when given alone. Pharmacol Toxicol 84: 226–233.
Johansson C, Jackson DM, Svensson L (1997). Nitric oxide synthase inhibition blocks phencyclidine-induced behavioural effects on prepulse inhibition and locomotor activity in the rat. Psychopharmacology (Berl) 131: 167–173.
Johansson C, Magnusson O, Deveney AM, Jackson DM, Zhang J, Engel JA et al (1998). The nitric oxide synthase inhibitor, L-NAME, blocks certain phencyclidine-induced but not amphetamine-induced effects on behaviour and brain biochemistry in the rat. Prog Neuropsychopharmacol Biol Psychiat 22: 1341–1360.
Kano T, Shimizu-Sasamata M, Huang PL, Moskowitz MA, Lo EH (1998). Effects of nitric oxide synthase gene knockout on neurotransmitter release in vivo. Neuroscience 86: 695–699.
Karatinos J, Rosse RB, Deutsch SI (1995). The nitric oxide pathway: potential implications for treatment of neuropsychiatric disorders. Clin Neuropharmacol 18: 482–499.
Kelly PH, Iversen SD (1976). Selective 6OHDA-induced destruction of mesolimbic dopamine neurons: abolition of psychostimulant-induced locomotor activity in rats. Eur J Pharmacol 40: 45–56.
Klamer D, Engel JA, Svensson L (2001). The nitric oxide synthase inhibitor, L-NAME, blocks phencyclidine-induced disruption of prepulse inhibition in mice. Psychopharmacology (Berl) 156: 182–186.
Klamer D, Engel JA, Svensson L (2004a). The neuronal selective nitric oxide synthase inhibitor, nomega-propyl-L-arginine, blocks the effects of phencyclidine on prepulse inhibition and locomotor activity in mice. Eur J Pharmacol 503: 103–107.
Klamer D, Engel JA, Svensson L (2004b). Phencyclidine-induced behaviour in mice prevented by methylene blue. Basic Clin Pharmacol Toxicol 94: 65–72.
Klamer D, Engel JA, Svensson L (2005a). Effects of phencyclidine on acoustic startle and prepulse inhibition in neuronal nitric oxide synthase deficient mice. Eur Neuropsychopharmacol 15: 587–590.
Klamer D, Palsson E, Fejgin K, Zhang J, Engel JA, Svensson L (2005b). Activation of a nitric-oxide-sensitive cAMP pathway with phencyclidine: elevated hippocampal cAMP levels are temporally associated with deficits in prepulse inhibition. Psychopharmacology (Berl) 179: 479–488.
Klamer D, Palsson E, Revesz A, Engel JA, Svensson L (2004c). Habituation of acoustic startle is disrupted by psychotomimetic drugs: differential dependence on dopaminergic and nitric oxide modulatory mechanisms. Psychopharmacology (Berl) 176: 440–450.
Laitinen JT, Laitinen KS, Tuomisto L, Airaksinen MM (1994). Differential regulation of cyclic GMP levels in the frontal cortex and the cerebellum of anesthetized rats by nitric oxide: an in vivo microdialysis study. Brain Res 668: 117–121.
Laitinen KS, Salovaara K, Severgnini S, Laitinen JT (1997). Regulation of cyclic GMP levels in the rat frontal cortex in vivo: effects of exogenous carbon monoxide and phosphodiesterase inhibition. Brain Res 755: 272–278.
Linn GS, Negi SS, Gerum SV, Javitt DC (2003). Reversal of phencyclidine-induced prepulse inhibition deficits by clozapine in monkeys. Psychopharmacology (Berl) 169: 234–239.
Luby ED, Cohen BD, Rosenbaum G, Gottlieb LS, Kelly R (1959). Study of a new schizophrenomimetic drug—sernyl. Arch Neurol Psychiatry 81: 363–369.
Mansbach RS, Geyer MA (1989). Effects of phencyclidine and phencyclidine biologs on sensorimotor gating in the rat. Neuropsychopharmacology 2: 299–308.
McGhie A, Chapman J (1961). Disorders of attention and perception in early schizophrenia. Br J Med Psychol 34: 103–116.
Moghaddam B, Adams B, Verma A, Daly D (1997). Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci 17: 2921–2927.
Palsson E, Fejgin K, Wass C, Engel JA, Svensson L, Klamer D (2007). The amino acid L-lysine blocks the disruptive effect of phencyclidine on prepulse inhibition in mice. Psychopharmacology (Berl) 192: 9–15.
Pepicelli O, Raiteri M, Fedele E (2004). The NOS/sGC pathway in the rat central nervous system: a microdialysis overview. Neurochem Int 45: 787–797.
Perry W, Geyer MA, Braff DL (1999). Sensorimotor gating and thought disturbance measured in close temporal proximity in schizophrenic patients. Arch Gen Psychiatry 56: 277–281.
Pifarre P, Garcia A, Mengod G (2007). Species differences in the localization of soluble guanylyl cyclase subunits in monkey and rat brain. J Comp Neurol 500: 942–957.
Prast H, Philippu A (2001). Nitric oxide as modulator of neuronal function. Prog Neurobiol 64: 51–68.
Reif A, Herterich S, Strobel A, Ehlis AC, Saur D, Jacob CP et al (2006). A neuronal nitric oxide synthase (NOS-I) haplotype associated with schizophrenia modifies prefrontal cortex function. Mol Psychiatry 11: 286–300.
Rothman RB, Reid AA, Monn JA, Jacobson AE, Rice KC (1989). The psychotomimetic drug phencyclidine labels two high affinity binding sites in guinea pig brain: evidence for N-methyl-D-aspartate-coupled and dopamine reuptake carrier-associated phencyclidine binding sites. Mol Pharmacol 36: 887–896.
Santiago M, Westerink BH (1990). Characterization of the in vivo release of dopamine as recorded by different types of intracerebral microdialysis probes. Naunyn Schmiedebergs Arch Pharmacol 342: 407–414.
Schneider M, Koch M (2005). Behavioral and morphological alterations following neonatal excitotoxic lesions of the medial prefrontal cortex in rats. Exp Neurol 195: 185–198.
Schwabe K, Koch M (2004). Role of the medial prefrontal cortex in N-methyl-D-aspartate receptor antagonist induced sensorimotor gating deficit in rats. Neurosci Lett 355: 5–8.
Seeman P, Lasaga M (2005). Dopamine agonist action of phencyclidine. Synapse 58: 275–277.
Seeman P, Ko F, Tallerico T (2005). Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Mol Psychiatry 10: 877–883.
Segovia G, Mora F (1998). Role of nitric oxide in modulating the release of dopamine, glutamate, and GABA in striatum of the freely moving rat. Brain Res Bull 45: 275–279.
Shinkai T, Ohmori O, Hori H, Nakamura J (2002). Allelic association of the neuronal nitric oxide synthase (NOS1) gene with schizophrenia. Mol Psychiatry 7: 560–563.
Smith JC, Whitton PS (2000). Nitric oxide modulates N-methyl-D-aspartate-evoked serotonin release in the raphe nuclei and frontal cortex of the freely moving rat. Neurosci Lett 291: 5–8.
Smith JC, Whitton PS (2001). The regulation of NMDA-evoked dopamine release by nitric oxide in the frontal cortex and raphe nuclei of the freely moving rat. Brain Res 889: 57–62.
Southam E, Garthwaite J (1993). The nitric oxide-cyclic GMP signalling pathway in rat brain. Neuropharmacology 32: 1267–1277.
Srivastava N, Barthwal MK, Dalal PK, Agarwal AK, Nag D, Srimal RC et al (2001). Nitrite content and antioxidant enzyme levels in the blood of schizophrenia patients. Psychopharmacology (Berl) 158: 140–145.
Suzuki E, Nakaki T, Nakamura M, Miyaoka H (2003). Plasma nitrate levels in deficit versus non-deficit forms of schizophrenia. J Psychiatry Neurosci 28: 288–292.
Swerdlow NR, Lipska BK, Weinberger DR, Braff DL, Jaskiw GE, Geyer MA (1995). Increased sensitivity to the sensorimotor gating-disruptive effects of apomorphine after lesions of medial prefrontal cortex or ventral hippocampus in adult rats. Psychopharmacology (Berl) 122: 27–34.
Takahata R, Moghaddam B (2003). Activation of glutamate neurotransmission in the prefrontal cortex sustains the motoric and dopaminergic effects of phencyclidine. Neuropsychopharmacology 28: 1117–1124.
Taneli F, Pirildar S, Akdeniz F, Uyanik BS, Ari Z (2004). Serum nitric oxide metabolite levels and the effect of antipsychotic therapy in schizophrenia. Arch Med Res 35: 401–405.
Vesely DL (1979). Phencyclidine stimulates guanylate cyclase activity. Biochem Biophys Res Commun 88: 1244–1248.
Wass C, Archer T, Palsson E, Fejgin K, Alexandersson A, Klamer D et al (2006a). Phencyclidine affects memory in a nitric oxide-dependent manner: working and reference memory. Behav Brain Res 174: 49–55.
Wass C, Archer T, Palsson E, Fejgin K, Klamer D, Engel JA et al (2006b). Effects of phencyclidine on spatial learning and memory: Nitric oxide-dependent mechanisms. Behav Brain Res 171: 147–153.
Wegener G, Volke V, Rosenberg R (2000). Endogenous nitric oxide decreases hippocampal levels of serotonin and dopamine in vivo. Br J Pharmacol 130: 575–580.
Wiley JL (1998). Nitric oxide synthase inhibitors attenuate phencyclidine-induced disruption of prepulse inhibition. Neuropsychopharmacology 19: 86–94.
Wiley JL, Kogut CP, Witherell JR (1999). Effects of acute and repeated dosing with PCP on locomotion in nNOS knockout mice. 29th Annual Meeting. Society for Neuroscience (Miami Beach: Society for Neuroscience). pp 946.
Wynn JK, Sergi MJ, Dawson ME, Schell AM, Green MF (2005). Sensorimotor gating, orienting and social perception in schizophrenia. Schizophr Res 73: 319–325.
Xing G, Chavko M, Zhang LX, Yang S, Post RM (2002). Decreased calcium-dependent constitutive nitric oxide synthase (cNOS) activity in prefrontal cortex in schizophrenia and depression. Schizophr Res 58: 21–30.
Yesavage JA, Freman AM (1978). Acute phencyclidine (PCP) intoxication: psychopathology and prognosis. J Clin Psychiatry 39: 664–666.
Yilmaz N, Herken H, Cicek HK, Celik A, Yurekli M, Akyol O (2007). Increased levels of nitric oxide, cortisol and adrenomedullin in patients with chronic schizophrenia. Med Princ Pract 16: 137–141.
Acknowledgements
This study was supported by grants from the Swedish Medical Research Council (4247), Stiftelsen Tornspiran, the Theodore and Vada Stanley Foundation, Göteborgs Läkaresällskap, Wilhelm och Martina Lundgrens Vetenskapsfond, Jubiluemsfonden vid Göteborgs Universitet, the Swedish Society of Medicine, Åke Wibergs Stiftelse, Adlerbertska Forskningsstiftelsen, Åhlén-Stiftelsen, the Swedish Society for Medical Research and Svenska Lundbeckstiftelsen. The technical assistance of Gun Andersson and Kenn Johannessen is gratefully acknowledged.
Author information
Authors and Affiliations
Corresponding author
Additional information
Disclosure/Conflict of Interest
The authors hereby declare that no financial support or compensation has been received from any individual or corporate entity over the past 3 years for research or professional service and there are no personal financial holdings that could be perceived as constituting a potential conflict of interest.
Rights and permissions
About this article
Cite this article
Fejgin, K., Pålsson, E., Wass, C. et al. Nitric Oxide Signaling in the Medial Prefrontal Cortex is Involved in the Biochemical and Behavioral Effects of Phencyclidine. Neuropsychopharmacol 33, 1874–1883 (2008). https://doi.org/10.1038/sj.npp.1301587
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301587
Keywords
This article is cited by
-
Reactivation of Tert in the medial prefrontal cortex and hippocampus rescues aggression and depression of Tert−/− mice
Translational Psychiatry (2016)
-
Exploring metabolic pathway disruption in the subchronic phencyclidine model of schizophrenia with the Generalized Singular Value Decomposition
BMC Systems Biology (2011)
-
Inhibition of neuronal nitric oxide synthase prevents alterations in medial prefrontal cortex excitability induced by repeated cocaine administration
Psychopharmacology (2011)
-
Effects of the Nitric Oxide Synthase Inhibitor L-NAME on Recognition and Spatial Memory Deficits Produced by Different NMDA Receptor Antagonists in the Rat
Neuropsychopharmacology (2010)
-
Information processing deficits and nitric oxide signalling in the phencyclidine model of schizophrenia
Psychopharmacology (2010)
