
Abstract
A sudden loud sound evokes an electromyographic (EMG) response from the orbicularis oculi muscle in humans together with an auditory evoked potential (AEP) and an increase in skin conductance (SC). Startle responses are inhibited by weak prepulses (prepulse inhibition, (PPI)) and may also be modified by the level of alertness. We compared the sedative drug clonidine and the alerting drug modafinil on sound-evoked EMG, AEP, and SC responses, on the PPI of these responses and on level of arousal and autonomic functions. Sixteen healthy male volunteers participated in four weekly sessions (clonidine 0.2 mg, modafinil 400 mg, their combination, placebo) in a double-blind, cross-over, balanced design. Responses were evoked by sound pulses of 115 and 85 dB (PPI) for 40 ms and recorded conventionally. Level of alertness, autonomic functions (pupil diameter, blood pressure, heart rate, salivation, temperature) and the plasma levels of the hormones prolactin, thyroid-stimulating hormone and growth hormone were also measured. Data were analyzed with analysis of variance with multiple comparisons. Both prepulses and clonidine attenuated all three startle responses and modafinil antagonized clonidine's effects on the EMG and AEP responses. None of the drugs affected PPI. Clonidine showed sedative and sympatholytic effects, and modafinil showed alerting and sympathomimetic effects. In conclusion, startle responses were susceptible not only to PPI but also to the level of arousal.
Similar content being viewed by others
INTRODUCTION
The startle response involves rapid involuntary contractions of facial and skeletal musculature in response to a sudden intense stimulus. In the case of the acoustic startle response, this reaction occurs in response to a sudden loud sound and is measurable in humans as an electromyographic (EMG) response from the orbicularis oculi muscle (eyeblink startle response). This response is believed to be mediated via a pathway below the level of the diencephalon consisting of between three and five central synapses, originating from the sensory receptors and terminating on the motor neurons, where the principal relay is the caudal pontine reticular nucleus (Davis et al, 1982; for review, see Yeomans and Frankland, 1996; Koch, 1999). In addition to this muscular response, changes in electroencephalographic (EEG) activity can be observed in response to the auditory stimulus, and these changes are termed auditory evoked potentials (AEP). In particular, the N1/P2 component of the AEP is measurable following the presentation of the acoustic startle stimulus (for examples, see Abduljawad et al, 1999, 2001; Phillips et al, 2000a, 2000b; Graham et al, 2001, 2004; Scaife et al, 2005, 2006). Both the muscular and the AEP responses are fast responses; longer-latency responding to the startle stimulus can be observed in autonomic activity, in particular in sympathetically mediated skin conductance. The skin conductance response (SCR) is an increase in conductance maximal approximately 6 s following the presentation of the stimulus (Turpin et al, 1999; Graham et al, 2005; Scaife et al, 2005).
The presentation of a brief, low-intensity acoustic stimulus 30–500 ms before the presentation of the loud, startle-eliciting stimulus attenuates the amplitude of the muscular startle response, a phenomenon termed prepulse inhibition (PPI; Graham, 1975; Davis, 1980; Swerdlow et al, 1992; Koch, 1999). The AEP response is also affected by PPI (Abduljawad et al, 1999; Phillips et al, 2000a, 2000b; Graham et al, 2004). PPI of the AEP response may have a neural mechanism that is different from the mechanism underlying PPI of the muscular response because the two responses show different pharmacological sensitivities (eg bromocriptine (Abduljawad et al, 1999), amitryptyline (Phillips et al, 2000a), acute tryptophan depletion (Phillips et al, 2000b), and clozapine (Graham et al, 2001)). PPI is thought to represent a form of ‘gating’, in which the influence of irrelevant or intrusive stimuli on ongoing behavior is prevented (Swerdlow et al, 1992; Koch, 1999). There are currently no reports on the effect of a prepulse on the SCR.
Apart from PPI, the acoustic startle response may also be modulated by the level of arousal. Indeed, it has been reported that the EMG startle reflex amplitude can be reduced by sedative drugs of different classes, for example, benzodiazepines (diazepam: Scaife et al, 2005), 5HT2 receptor antagonists (ketanserin: Graham et al, 2002), α2-adrenoceptor agonists (clonidine: Kumari et al, 1996; Abduljawad et al, 1997, 2001), tricyclic antidepressants (amitriptyline: Phillips et al, 2000a), and atypical antipsychotics (clozapine and quetiapine: Graham et al, 2001, 2004). In contrast to the EMG startle response, the AEP is relatively resistant to sedative drugs, with only the benzodiazepines reducing N1/P2 amplitude consistently (Lader, 1977; Bond et al, 1983; Pooviboonsuk et al, 1996; Curran et al, 1998; Abduljawad et al, 2001; Scaife et al, 2005). However, no information is available on the effects of sedative drugs on SCR.
Because sedative drugs consistently suppress the EMG startle response, it could be expected that alerting drugs could have an opposite effect, that is, they would enhance startle response amplitude. There is some indication that this may be the case, as it has been reported that the psychostimulant amphetamine enhances the acoustic startle response (Davis et al, 1975; Kokkinidis and MacNeill, 1982; Swerdlow et al, 1990). However, it is not known whether other alerting drugs share this effect. Modafinil is a novel wakefulness-promoting drug used in the treatment of narcolepsy (US Modafinil in Narcolepsy Multicentre Study Group, 1998, 2000). Although the mode of action of modafinil is currently unknown, it has been suggested that it interacts with the central noradrenergic arousal pathway involving the locus coeruleus (LC) (Saper and Scammell, 2004; Saper et al, 2005) and also with dopaminergic mechanisms involved in the regulation of alertness (Mignot et al, 1994; Nishino et al, 1998; Wisor et al, 2001; Wisor and Eriksson, 2005). Clonidine is an α2-adrenoceptor agonist with potent sedative effects that may be related to its inhibitory action on the LC (De Sarro et al, 1987). It has been shown previously that modafinil and clonidine influence the level of alertness and sympathetic functions in opposite directions (Hou et al, 2005), consistent with their interactions with central noradrenergic mechanisms.
In the present study we compared the effects of single doses of modafinil and clonidine on all three startle responses (EMG, AEP, SCR), as well as PPI of those responses, in a group of healthy male volunteers. We also examined the possible functional antagonistic interaction between the sedative and alerting drug on all these measures. The effects of the treatments were monitored on quantitative measures of arousal together with autonomic and endocrine functions, which are influenced by central noradrenergic mechanisms (Hou et al, 2005; Samuels et al, 2006).
MATERIALS AND METHODS
Subjects
Sixteen healthy male volunteers aged 18–45 years (mean±standard error of mean (SEM): 22.7±1.6 years), 171–190 cm (mean±SEM: 181.4±1.45 cm) in height and weighing 56.4–92.9 kg (mean±SEM: 74.7±2.8 kg), with a hearing threshold of 5–19 dB (mean±SEM: 6.9±0.9 dB), participated in the study. Subjects were all medication free for at least 3 months before the start of the study and completed a brief medical history and physical examination before inclusion in the study. Volunteers were all non-smokers and were asked to avoid drinking alcohol, coffee, and other caffeine-containing beverages for at least 24 h before each experimental session and to avoid taking any medication for the duration of the study. The study protocol was approved by the University of Nottingham Medical School Ethics Committee, and all volunteers gave their written consent after reading a detailed information sheet.
Drugs
Modafinil (400 mg), clonidine (0.2 mg), modafinil (400 mg)+clonidine (0.2 mg), and placebo were administered orally in matching capsules for double-blind administration. The doses were chosen on the basis of the current literature (modafinil: Taneja et al, 2005; clonidine: Morley et al, 1991; Bitsios et al, 1996; Phillips et al, 2000c, 2000d). The time required to attain peak plasma concentrations (tMAX) of clonidine and modafinil after the ingestion of single doses is approximately 2 h (modafinil: Wong et al, 1998; clonidine: Lowenthal et al, 1988), and the time course of the sessions was designed to ensure peak plasma concentrations of the drugs at the start of the post-treatment testing.
Design
Subjects participated in four sessions at weekly intervals, returning to the laboratory at the same time each week. Subjects were allocated to drug conditions according to a double-blind, balanced, cross-over design. The time course of the sessions was designed with regard to the pharmacokinetic profile of the two active drugs (see above).
Tests and Apparatus
Acoustic startle response
The method used was similar to that of Abduljawad et al (2001). All recordings were made while the subject was seated in an armchair.
A Kamplex AC30 Clinical Audiometer (P.C. Werth Ltd, London, UK) was used to generate binaural auditory stimuli; a background tone of 70 dB [A] at 1 kHz was present throughout, and sound pulses of 115 [A] or 85 dB [A] at 1 kHz were presented for 40 ms at varying intervals. The period of recording started with a 60-s adaptation period in which only the background sound was presented. This was followed by a single 115-dB [A] pulse, the responses to which were discarded. The remainder of the recording period consisted of 24 trials separated by varying inter-trial intervals of between 15 and 35 s, with a mean inter-trial interval of 25 s. The two stimulus combinations presented in the trials were (i) a single 40-ms 115-dB [A] pulse (12 pulse-alone trials) and (ii) a 40-ms 85-dB [A] pulse followed by a 40-ms 115-dB [A] pulse with an inter-stimulus interval of 120 ms (12 prepulse/pulse trials). The order of presentation of the two types of trial was randomized, with the constraint that the same type of trial did not occur more than three times in succession.
A CED 1401+computer with a 1902 interface (Cambridge Electronic Design Ltd, Cambridge, UK) was used to record the EMG response of the right orbicularis oculi muscle to the presentation of the acoustic stimuli. The response was measured using two silver/silver chloride disc electrodes of 0.5-cm diameter placed on the lower eyelid with a ground electrode placed over the right mastoid. The EMG data were rectified using a 1-Hz high-pass filter with a notch filter set at 50 Hz to minimize mains electrical interference.
Auditory evoked potential
A second CED 1902 interface was used to obtain single-channel recordings of the EEG response, the AEP, to the presentation of the acoustic stimuli simultaneously with the EMG recordings. The response was measured using a 0.5-cm silver/silver chloride disc electrode placed on the scalp at the Cz (vertex) position and an electrode pad placed on the left mastoid. The ground electrode was placed on the bridge of the nose. The vertex potentials were displayed in a ‘positive up’ configuration.
Skin conductance response
A CED 2502 interface connected to the same 1401+computer used to record EMG and EEG responses was used to record the SCR to the presentation of the acoustic stimuli. The response was measured using electrode pads on the terminal phalanges of the first and third digits of the right hand.
Measures of alertness
The Leeds Psychomotor Tester (Psychopharma Ltd, Surrey, UK) was used to collect critical flicker fusion frequency (CFFF) measurements, defined as the frequency at which a flickering light appears to be continuous (Smith and Misiak, 1976). The CFFF test was conducted conventionally, with eight threshold measurements collected per session: four with increasing frequencies and four with decreasing frequencies. The mean of the eight measurements was taken as the value of the CFFF (see Samuels et al, 2006).
A computerized battery of 16 visual analogue scales (VASs) was used to collect self-ratings of alertness, calmness, and contentedness. Sixteen contrasting statements were rated along a continuous 10-cm line to represent the participant's subjective emotional and physiological state, for example, ‘happy’ vs ‘sad’ and ‘alert’ versus ‘drowsy’ (Norris, 1971; Bond and Lader, 1974; see Samuels et al, 2006). The position of the poles (left or right) and the order of presentation of the scales were randomized between subjects and sessions.
A dedicated monocular television pupillometer (setup version 1.20: AMtech, Weinheim, Germany) was used to collect the pupillographic sleepiness test (PST) measurements, defined as the spontaneous pupillary fluctuations in darkness over an 11-min period. These pupil fluctuations are regarded as a physiological index of alertness level (Lowenstein et al, 1963; Yoss et al, 1970; Lüdtke et al, 1998). The PST quantitatively analyzes these pupil fluctuations and yields two measurements: pupillary unrest index (PUI: the distance traveled by the margin of the pupil over a 1-min period) and the total power of the pupil diameter fluctuations (obtained from a fast Fourier transform). Pupil diameter was measured continuously at a frequency of 25 Hz (see Samuels et al, 2006).
Autonomic functions
A binocular infra-red video pupillometer (Procyon Ltd, London, UK) with a calibrated internal light source was used to obtain resting pupil diameter measurements (static pupillometry). Pupillometry was conducted in a darkened room in darkness and at three luminance levels (6, 91, and 360 cd m−2), with each measurement recorded over 2 s at 4 Hz (see Samuels et al, 2006).
An electroaneroid sphygmomanometer was used to record blood pressure and heart rate in both standing and supine positions. A Braun Pro4000 tympanic thermometer (Welch Allyn UK Ltd, Buckinghamshire, UK) was used to measure temperature from the ear canal. A measure of salivation was derived by placing three cotton wool dental rolls in the subject's mouth (two buccally and one sublingually) and recording the increase in their weight over a 1-min period (Peck, 1959; Arya et al, 1997; Szabadi and Tavernor, 1999). The test was repeated twice with a 5-min interval, and the mean of the two measurements was taken as an index of salivary output.
Endocrine functions
A 10-ml blood sample was taken from an antecubital vein and analyzed for concentrations of the hormones prolactin and thyroid-stimulating hormone (TSH) by enzyme immunoassay and for growth hormone (GH) by chemiluminescence immunoassay in the Clinical Chemistry Laboratory (Queen's Medical Centre, Nottingham, UK).
Procedure
After a 15-min acclimatization period, subjects completed 30 min of pretreatment testing, including standing and supine heart rate and blood pressure, temperature, salivation, CFFF, VAS, PST, and resting pupil diameter. Two hours after ingestion of the capsule the post-treatment tests were conducted over 45 min, including standing and supine heart rate and blood pressure, temperature, salivation, CFFF, VAS, PST, resting pupil diameter, acoustic startle paradigm, and blood sampling.
Data Analysis and Statistics
The EMG responses recorded in each type of trial were averaged across the 12 trials using Spike 2 software (Cambridge Electronic Design Ltd, Cambridge, UK). The latencies (time taken from the onset of the stimulus to the onset of the response, in seconds), rise times (time taken from the onset of the response to the peak, in seconds) and amplitudes (maximal change in EMG potential, in millivolts) of the responses to the pulse alone and the prepulse/pulse were obtained from the averaged responses and used for further analysis. Percentage PPI was calculated for further analysis using the formula

where Apulse alone and Aprepulse/pulse are the amplitudes of the mean responses to the 115-dB pulses in the pulse-alone and prepulse/pulse trials, respectively (Abduljawad et al, 2001). An example EMG trace is shown in Figure 1 (top).

Examples of the averaged responses recorded over 12 pulse-alone trials (left-hand panels) and 12 prepulse/pulse trials (right-hand panels) for the acoustic startle response (a), the N1/P2 complex of the AEP (b), and the SCR (c). Stimulus presentation is represented by a black rectangle. The measurements derived from these recordings are labeled. The response amplitudes measured from these trials were used to calculate percentage PPI of the three responses (see text for details).
The EEG AEPs recorded in each type of trial were averaged across the 12 trials (as EMG, above). The time to the N1 and P2 components of the response (in seconds) and the amplitude of the N1/P2 complex (in millivolts; taken as the amplitude difference from the peak of the N1 wave to the peak of the P2 wave, Mauguière et al, 1995) were obtained from the averaged responses to the pulse alone and the prepulse/pulse and were used for further analysis. The range of latencies to the peak of the wave following stimulus onset was taken as 81–140 ms for N1 and 141–240 ms for P2. Percentage PPI of the N1/P2 complex was calculated using the formula stated above (Abduljawad et al, 2001). A sample EEG trace is shown in Figure 1 (center).
The SCRs recorded in each type of trial, measured from the onset of the response to the maximum conductance achieved within 10 s of the stimulus delivery (see Scaife et al, 2005), were averaged across the 12 trials (as EMG, above). The latencies (time taken from the onset of the stimulus to the onset of the response, in seconds), rise times (time taken from the onset of the response to the peak, in seconds), and amplitudes (maximal change in skin conductance, in micro-siemens) of the responses to the pulse alone and prepulse/pulse were obtained from the averaged responses and used for further analysis. Percentage PPI of the amplitude was calculated using the formula stated above (Abduljawad et al, 2001). A sample SCR trace is shown in Figure 1 (bottom).
Self-rated values of ‘alertness’, ‘calmness’, and ‘contentedness’ were derived from the VAS scores after weighting on these factors (Bond and Lader, 1974). Pretreatment/post-treatment differences for these ratings and for the CFFF values were calculated for further analysis. Pretreatment/post-treatment differences were also calculated for the PST parameters of power, PUI and pupil diameter, and these values were used for statistical analysis.
Pretreatment/post-treatment differences were analyzed for all autonomic measures with the exception of resting pupil diameter at different luminance levels, where the pretreatment and post-treatment data were analyzed separately. Pretreatment/post-treatment differences were not calculated for these pupillary measurements because the recordings were taken at different luminance levels, and calculating the difference would have eliminated the effect of luminance on the measures studied. All pupil data were averaged across the left and right eyes.
Effects of the treatments on the secretion of prolactin, TSH, and GH were analyzed using post-treatment plasma concentration values.
The data were initially checked for skew and subjected to a transformation where indicated. All data were analyzed using within-subject analysis of variance (ANOVA). Pretreatment values were analyzed to find any session effects within the results. No significant pretreatment session effects were found, and so pretreatment/post-treatment differences were taken as the dependent variable where appropriate (see above). Two-way within-subject ANOVA was used to analyze the EMG, AEP, and SCR data (drug condition, four levels; pulse condition, two levels) and resting pupil diameter data (drug condition, four levels; luminance level, four levels). Because the primary measure of interest was the effect of treatment condition on the acoustic startle response evoked by the presentation of the pulse alone, one-way within-subject ANOVA was conducted on pulse-alone data subsequent to the detection of a significant effect in the two-way ANOVA. One-way within-subject ANOVA (drug condition, four levels) was used to analyze the alertness, autonomic and endocrine measures, and PPI data. All significant main effects were further analyzed using Dunnett's corrected t-test (df=45, k=4): active treatment conditions were compared with placebo (criterion of significance p<0.05).
RESULTS
All pretreatment values were initially analyzed using one-way ANOVA to determine the presence of any session effects. No significant effects were found within the pretreatment data (p<0.05) and so pretreatment/post-treatment differences were taken as the dependent measure where appropriate (see above).
Acoustic Startle Response
The effects of the four treatment conditions on the three measures of EMG response (latency, rise time, amplitude) to the single pulse and the PPI of the amplitude are shown in Figure 2. Owing to technical reasons, one subject was excluded from the analysis of rise time, amplitude, and PPI. For latency, the two-way ANOVA (log10 transformation) showed an effect of drug condition (F3,45=3.88, p<0.05) and pulse condition (F1,15=5.83, p<0.05) but no significant interaction (F3,45=0.78, NS). The effect of pulse condition was due to a reduction in latency to respond in the presence of the prepulse compared to the presentation of the pulse alone. A one-way ANOVA (log10 transformation) of drug condition on the pulse-alone data showed a significant effect of treatment (F3,45=5.72, p<0.01), in which clonidine increased the latency of the EMG startle response. For rise time, the two-way ANOVA (square root transformation) showed no effect of pulse condition (F1,14=0.56, NS) or drug condition (F3,42=1.16, NS) and no significant interaction (F3,42=0.54, NS). For amplitude, the two-way ANOVA (square root transformation) showed effects of both pulse condition (F1,15=13.25, p<0.01) and drug condition (F3,45=2.81, p=0.05) but no significant interaction (F3,45=2.30, NS). The effect of pulse condition was due to suppression of amplitude in the presence of the prepulse compared to the presentation of the pulse alone. A one-way ANOVA (square root transformation) of drug condition on the pulse-alone data showed a significant effect of treatment (F3,45=3.45, p<0.05), in which clonidine reduced the amplitude of the startle response. There was no effect of treatment on the PPI of the amplitude (square root transformation; F3,42=0.73, NS).

EMG responses of orbicularis oculi muscle to the 115-dB pulses in the pulse-alone trials: latency (s; top left), rise time (s; top right), amplitude (mV; bottom left) and PPI of the amplitude (%; bottom right). Columns correspond to mean values of the parameters for the group (n=16); vertical bars represent SEM. Treatment condition is indicated at the bottom of the graphs: Pl=placebo, Clon=0.2 mg clonidine, Mod=400 mg modafinil, Combi=combination of 0.2 mg clonidine and 400 mg modafinil. Clonidine significantly increased the latency and reduced the amplitude of the startle response: *p<0.05 (Dunnett's test: comparison with placebo condition). The response amplitude was significantly reduced following a weak prepulse (PPI; see text for details).
Auditory Evoked Potential
The effects of the four treatment conditions on the three measures of the AEP response (latency to N1 peak, latency to P2 peak, N1/P2 amplitude) and the PPI of the amplitude are shown in Figure 3. Owing to technical reasons, two subjects were excluded from the analysis of these measures. For latency to N1 peak, the two-way ANOVA showed a trend for an effect of drug condition (F3,39=2.83, p=0.051) but no effect of pulse condition (F1,13=0.92, NS) and no significant interaction (F3,39=1.83, NS). A one-way ANOVA of drug condition on the pulse-alone data showed a significant effect of treatment (F3,39=4.49, p<0.05), in which clonidine increased the time to the N1 startle response. For latency to P2 peak, the two-way ANOVA showed an effect of pulse condition (F1,13=40.45, p<0.001) and drug condition (F3,39=5.64, p<0.05) but no significant interaction (F3,39=1.47, NS). The effect of pulse condition was due to a reduction in time to reach the P2 peak in the presence of the prepulse compared with the presentation of the pulse alone. A one-way ANOVA of drug condition on the pulse-alone data showed a significant effect of treatment (F3,39=3.96, p<0.05), in which modafinil reduced the latency to the P2 peak. For N1/P2 amplitude, the two-way ANOVA showed effects of both pulse condition (F1,13=86.74, p<0.001) and drug condition (F3,39=3.08, p<0.05) but no significant interaction (F3,39=2.67, NS). The effect of pulse condition was due to an attenuation of the N1/P2 amplitude in the presence of the prepulse compared with the presentation of the pulse alone. A one-way ANOVA of drug condition on the pulse-alone data showed a significant effect of treatment (log10 transformation; F3,39=4.51, p<0.01), in which clonidine reduced the amplitude of the N1/P2 complex. There was no effect of treatment on the PPI of the amplitude (log10 transformation; F3,39=1.62, NS).

Auditory evoked potential evoked by 115-dB pulses in the pulse-alone trials: latency to N1 peak (s; top left), latency to P2 peak (s; top right), N1/P2 amplitude (mV; bottom left), and PPI of the amplitude (%; bottom right). Columns correspond to mean values of the parameters for the group (n=14; data from two subjects were discarded for technical reasons); vertical bars represent SEM. Treatment condition is indicated at the bottom of the graphs: Pl=placebo, Clon=0.2 mg clonidine, Mod=400 mg modafinil, Combi=combination of 0.2 mg clonidine and 400 mg modafinil. Clonidine significantly increased the latency to N1 peak and reduced the N1/P2 amplitude; modafinil significantly reduced the latency to P2 peak: *p<0.05 (Dunnett's test: comparison with placebo condition). The response amplitude was significantly reduced following a weak prepulse (PPI; see text for details).
Skin Conductance Response
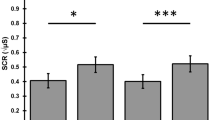
The effects of the four treatment conditions on the three measures of the SCR (latency, rise time, amplitude) and the PPI of the amplitude are shown in Figure 4. For latency, the two-way ANOVA showed no effects of drug condition (F3,45=0.79, NS) or pulse condition (F1,15=1.31, NS) and no interaction (F3,45=2.02, NS). For rise time, the two-way ANOVA showed an effect of pulse condition (F3,45=5.05, p<0.05) and a trend for an effect of drug condition (F1,15=2.37, p=0.08), but no significant interaction (F3,45=0.29, NS). The effect of pulse condition was due to a reduction in the time to response peak in the presence of the prepulse compared to the presentation of the pulse alone. A one-way ANOVA of drug condition on the pulse-alone data showed no effect of treatment compared to placebo (F3,45=2.11, NS). For amplitude, the two-way ANOVA showed an effect of pulse condition (F1,15=16.15, p=0.001) and drug condition (F3,45=7.38, p<0.001) but no significant interaction (F3,45=0.52, NS). The effect of pulse condition was due to an attenuation of amplitude in the presence of the prepulse compared to the presentation of the pulse alone. A one-way ANOVA of drug condition on the pulse-alone data showed a significant effect of treatment (F3,45=8.48, p<0.001), in which both clonidine and the combination of clonidine and modafinil reduced the amplitude of the SCR. There was no effect of treatment on the PPI of the amplitude (log10 transformation; F3,45=0.45, NS).

SCR evoked by 115-dB pulses in the pulse-alone trials: latency (s; top left), rise time (s; top right), amplitude (μS; bottom left) and PPI of the amplitude (%; top right). Columns correspond to mean values of the parameters for the group (n=16); vertical bars represent SEM. Treatment condition is indicated at the bottom of the graphs: Pl=placebo, Clon=0.2 mg clonidine, Mod=400 mg modafinil, Combi=combination of 0.2 mg clonidine and 400 mg modafinil. Clonidine and the combination of clonidine and modafinil significantly reduced the amplitude of the startle response: *p<0.05 (Dunnett's test: comparison with placebo condition). The response amplitude was significantly reduced following a weak prepulse (PPI; see text for details).
Alertness
Critical flicker fusion frequency
The effects of the four treatment conditions on the CFFF measurements are shown in Table 1. There was a significant effect of treatment (F3,45=56.44, p<0.001), in which CFFF was reduced following both clonidine administration and the combination of clonidine and modafinil. The apparent increase in CFFF following modafinil just failed to reach significance.
Visual analogue scales
The effects of the four treatment conditions on the three VAS factors are shown in Table 1. There was a significant effect of treatment on alertness (log10 transformation; F3,45=14.09, p<0.001), in which alertness was reduced following the administration of both clonidine and the combination of clonidine and modafinil and increased following the administration of modafinil. The calmness factor showed a significant effect of treatment (F3,45=3.56, p<0.05), in which calmness was reduced following modafinil administration. The contentedness factor showed a significant effect of treatment (log10 transformation, F3,45=4.89, p<0.01), in which contentedness was reduced following the administration of clonidine and the combination of clonidine and modafinil.
Pupillographic sleepiness test
The effects of the four treatment conditions on the PUI, total power of pupillary fluctuations, and pupil diameter measurements from the PST are shown in Table 1. There was a significant effect of treatment on PUI (F3,45=20.73, p<0.001), in which PUI was increased by the administration of both clonidine and the combination of clonidine and modafinil and was decreased by the administration of modafinil. Similarly, there was a significant effect of treatment on the total power of pupillary fluctuations (F3,45=20.54, p<0.001), in which the administration of both clonidine and the combination of clonidine and modafinil increased the total power of the fluctuations, while the administration of modafinil reduced the total power. There was also a significant effect of treatment on pupil diameter (F3,45=45.22, p<0.001), in which the administration of both clonidine and the combination of clonidine and modafinil decreased pupil diameter. The apparent increase in pupil diameter following modafinil administration just failed to reach significance.
Autonomic Functions
Pupil diameter
Figure 5 shows the effect of treatment condition on pretreatment (top) and post-treatment (bottom) measures of pupil diameter in darkness and at three increasing luminance levels. A two-way ANOVA (treatment × luminance) on the pretreatment data reveals an effect of luminance (F3,45=190.01, p<0.001) but no effect of treatment (F3,45=0.14, NS) and no significant interaction (F39,135=1.44, NS). A two-way ANOVA (treatment × luminance) on the post-treatment data reveals effects of both luminance (F3,45=261.32, p<0.001) and treatment (F3,45=48.45, p<0.001) and a significant interaction (F9,135=6.04, p<0.001). Analysis of pupil diameter at each luminance level separately, using one-way ANOVA, demonstrated an effect of treatment in darkness (F3,45=17.17, p<0.001) and at 6 cd m−2 (F3,45=38.06, p<0.001), 91 cd m−2 (F3,45=33.12, p<0.001) and 360 cd m−2 (F3,45=30.70, p<0.001). Multiple comparisons showed that the treatment effects were due to pupillary constriction produced by clonidine and the combination of clonidine and modafinil at all luminance levels as well as pupillary dilatation produced by modafinil at 360 cd m−2.

Resting pupil diameter: relationship between level of luminance and pupil diameter pretreatment (top) and post-treatment (bottom). The four treatment conditions are indicated by different symbols (see inset). Ordinate: absolute pupil diameter (mm); abscissa: level of luminance (cd m−2). Each point corresponds to the mean obtained for the group (n=16). Vertical bars represent two standard errors of the difference (2SED) obtained from the interaction term of the ANOVA. Clonidine and the combination of clonidine and modafinil reduced pupil diameter at all luminance levels, *p<0.05; modafinil increased pupil diameter at 360 cd m−2, +p<0.05 (Dunnett's test: comparison with placebo condition).
Heart rate
The effects of the four treatment conditions on heart rate in the supine and standing conditions are shown in Table 2. There was no effect of treatment on measures of heart rate in the standing or in the supine position. There was a significant effect of treatment on the orthostatic change in heart rate (F3,45=5.80, p<0.05), in which clonidine administration increased this measure.
Blood pressure
The effects of the four treatment conditions on systolic and diastolic blood pressure in the supine and standing positions are shown in Table 2. There was a significant effect of treatment on systolic blood pressure in the supine (F3,45=17.36, p<0.001) and standing (F3,45=10.93, p<0.001) positions. The administration of clonidine reduced both supine and standing systolic blood pressure, while the administration of modafinil increased supine systolic blood pressure. The apparent increase in standing systolic blood pressure following modafinil just failed to reach significance. There was no effect on supine systolic blood pressure following the administration of the combination of clonidine and modafinil; standing systolic blood pressure was reduced following this treatment. There was a significant effect of treatment on diastolic blood pressure in the supine (F3,42=11.17, p<0.001) and standing (log10 transformation; F3,45=12.72, p<0.001) positions. Administration of clonidine and the combination of clonidine and modafinil reduced diastolic blood pressure in both the supine and the standing positions. The apparent increase in diastolic blood pressure in both the supine and the standing positions following modafinil administration failed to reach significance. There was no effect of treatment on orthostatic change for either systolic or diastolic blood pressure.
Temperature
The effects of the four treatment conditions on core temperature are shown in Table 2. There was a significant effect of treatment on temperature measurements (F3,45=5.48, p<0.05), in which the administration of the combination of clonidine and modafinil reduced core temperature.
Salivation
The effects of the four treatment conditions on salivation are shown in Table 2. There was a significant effect of treatment on salivation measurements (F3,45=15.22, p<0.001), in which the administration of both clonidine and the combination of clonidine and modafinil reduced salivation.
Endocrine Functions
The effects of the four treatment conditions on the plasma concentrations of prolactin, TSH, and GH are shown in Figure 6.

Post-treatment plasma concentrations of prolactin (left), TSH (center), and GH (right). Columns correspond to mean values of the parameters for the group (n=16); vertical bars represent SEM. Treatment condition is indicated at the bottom of the graphs: Pl=placebo, Clon=0.2 mg clonidine, Mod=400 mg modafinil, Combi=combination of 0.2 mg clonidine and 400 mg modafinil. The ordinate axis represents the hormone plasma concentration (mIUL−1). Clonidine significantly increased TSH concentration and modafinil significantly decreased prolactin concentration: *p<0.05 (Dunnett's test: comparison with placebo condition).
Prolactin
There was a significant effect of treatment condition on plasma prolactin levels (log10 transformation; F3,45=19.59, p<0.001), in which modafinil and the combination of clonidine and modafinil reduced the concentration of prolactin
TSH
There was a significant effect of treatment condition on plasma TSH levels (log10 transformation; F3,45=3.13, p<0.05), in which clonidine increased the concentration of TSH.
GH
There was no significant effect of treatment condition on plasma GH levels (log10 transformation; F3,42=0.53, NS).
DISCUSSION
In the present experiment, a single dose of 0.2 mg clonidine reduced both the subjective and the objective level of alertness in healthy volunteers (see Table 1), in agreement with previous reports (Morley et al, 1991; Kumari et al, 1996; Abduljawad et al, 1997, 2001; Phillips et al, 2000d; Hou et al, 2005, 2006). Alertness was robustly increased by a single dose of 400 mg modafinil, in contrast to previous reports using a 200-mg single dose where either a small effect (Hou et al, 2005, 2007) or no effect (Samuels et al, 2006) of modafinil in healthy non-sleep-deprived volunteers was observed. In the latter study it was suggested that a sleep-deprivation protocol would be necessary to observe alertness-enhancing effects in healthy volunteers, because modafinil at a dosage of 200 mg has been effective in promoting alertness when healthy volunteers were sleep-deprived (Wesensten et al, 2002; Walsh et al, 2004). It has also been found to be alerting in patients with reduced baseline alertness levels (US Modafinil in Narcolepsy Multicentre Study Group, 1998, 2000; Pack et al, 2001; Rammohan et al, 2002; Szabadi et al, 2002; Rosenthal and Bryant, 2004). However, the present study has demonstrated that the larger dose of 400 mg is sufficient for the observation of an alerting effect in a small group of healthy volunteers. Despite the larger dose of modafinil used and the robust alerting effects of the drug administered alone, the administration of modafinil in combination with clonidine failed to antagonize the sedative effects produced by clonidine alone. Thus, the sedative effect of clonidine 0.2 mg appears to supersede the alerting effect of 400 mg modafinil. In addition to the opposite effects of clonidine and modafinil on subjectively rated alertness, the two drugs also had differential effects on the other two factors rated on the VAS: calmness was reduced by modafinil and unaffected by clonidine, and contentedness was reduced by clonidine and unaffected by modafinil. The reduction in calmness in response to modafinil may indicate a mild anxiogenic effect of this relatively high dosage of the drug, although it was not observed using the lower dosage level of 200 mg (Samuels et al, 2006). The reduction of subjective contentedness by clonidine indicates that the experience of sedation may be perceived as unpleasant.
The presentation of the auditory pulse stimulus produced a startle response in the orbicularis oculi muscle that was altered by the administration of clonidine: the latency to respond to the stimulus was increased and the amplitude of the response was reduced (see Figure 2). The effect on response amplitude is in agreement with previous reports on the effect of clonidine on the acoustic startle response amplitude (Kumari et al, 1996; Abduljawad et al, 1997, 2001) and also with previous reports on the effect of sedative drugs on the startle response amplitude (Phillips et al, 2000a; Graham et al, 2001, 2002, 2004; Scaife et al, 2005). The effect on latency is consistent with a reduced startle response to the acoustic stimulus.
Clonidine is known to act on presynaptic α2 adrenoceptors in the LC and reduce neuronal activity (Szabadi and Bradshaw, 1996). The noradrenergic neurons of the LC, a major wakefulness-promoting nucleus, innervate the cerebral cortex (Jones et al, 1977; Robbins, 1984; Jones and Yang, 1985; Séguéla et al, 1990) and increase EEG signs of arousal (Berridge and Foote, 1991). Apart from directly activating the cerebral cortex, the LC also exerts an indirect alerting effect by inhibiting the ventrolateral preoptic nucleus (VLPO) of the hypothalamus (Nelson et al, 2002, 2003). It is possible that the sedative effects of certain drugs, including clonidine, may reflect an underlying reduction in the activity of the LC and thus a reduction in the arousal-enhancing outputs to the cortex. It is of interest that the LC also projects to the motoneurons in the facial nucleus (Jones and Yang, 1985), which innervates the orbicularis oculi muscle involved in the acoustic startle response, and that the application of noradrenaline enhances the excitatory activity of these motoneurons (VanderMaelen and Aghajanian, 1980; Rasmussen and Aghajanian, 1990; White et al, 1991). Thus, the LC has a facilitatory influence on the facial nucleus, enhancing the tone of the orbicularis oculi muscle. It is therefore an intriguing possibility that a reduction in LC activity brought about by sedative drugs leads to a reduction in the tone of the orbicularis oculi muscle manifesting as a reduction in EMG startle response amplitude. We suggest that the suppression of startle response amplitude by sedative drugs may be due to a reduction in LC activity leading to the attenuation of the noradrenergic facilitation of the facial nucleus.
If our hypothesis is correct, it would have been expected that drugs enhancing LC activity would enhance startle reflex amplitude. In support of this hypothesis, it has been reported that the α2 adrenoceptor antagonist yohimbine, which is known to enhance LC activity (Ivanov and Aston-Jones, 1995; Singewald and Sharp, 2000; Tanaka et al, 2000), enhances the amplitude of the acoustic startle response (Morgan et al, 1993). While modafinil on its own had no effect upon startle response latency or amplitude in the present experiment (see Figure 2), its administration in combination with clonidine resulted in an acoustic startle response that was not significantly different from the response obtained in the placebo condition. This indicates a functional antagonistic relationship between clonidine and modafinil, which in turn may reflect their opposite effects on LC activity (Hou et al, 2005).
The acoustic startle response amplitude was significantly suppressed by the presentation of a weak prepulse before the presentation of the startle-eliciting pulse, that is, there was significant PPI. This is in agreement with many previous reports (for example, Graham, 1975; Abduljawad et al, 1999; Phillips et al, 2000a, 2000b; Graham et al, 2001, 2002, 2004; Bitsios et al, 2005; for reviews see also Davis, 1980; Swerdlow et al, 1992; Koch, 1999). None of the treatment conditions had any significant effect on PPI of the acoustic startle response (see Figure 2). For clonidine, this is in agreement with previous reports in healthy volunteers in which no effects on PPI were found (Abduljawad et al, 1997, 2001). In this respect it is of interest that, whereas the α2 adrenoceptor agonist clonidine does not affect PPI in humans, the administration of α1-adrenoceptor agonists in rats (Carasso et al, 1998) and the absence of α2-adrenoceptors in transgenic mice (Sallinen et al, 1998) have been reported to lead to the disruption of PPI.
Like clonidine, modafinil was without any effect on PPI of the acoustic startle response. This negative finding is of special interest because the dopaminergic system has been implicated in the mode of action of modafinil and the dopaminergic system is known to play a key role in mediating PPI (Swerdlow et al, 1992; Koch and Schnitzler, 1997; Koch, 1999; Zhang et al, 2000). Indeed, it has been shown that amphetamine, a wakefulness-promoting drug acting via dopaminergic mechanisms (Bunney et al, 1973a, 1973b; Calcagnetti and Schechter, 1992; Nishino et al, 1998; Paladini et al, 2001), disrupts PPI of the startle response in both rat (Mansbach et al, 1988; Swerdlow et al, 1990; Sills, 1999; Zhang et al, 2000; Swerdlow et al, 2003; Brunell and Spear, 2006) and human (Kumari et al, 1998; Hutchison and Swift, 1999; Swerdlow et al, 2003) startle paradigms. Although both modafinil and amphetamine promote wakefulness, modafinil does not seem to share the psychostimulant and addictive potentials of amphetamine (Simon et al, 1995; Ferraro et al, 1997; Engber et al, 1998a, 1998b; Jasinski, 2000; Deroche-Gamonet et al, 2002; Chapotot et al, 2003), which are known to be mediated via the meso-accumbal dopaminergic system (Di Ciano et al, 1995; Zhang et al, 2000; Di Chiara et al, 2004). It is therefore tempting to speculate that modafinil may spare this dopaminergic projection while enhancing the activity of other dopaminergic pathways important in the maintenance of alertness. However, it should be noted that in studies in which amphetamine-induced PPI disruption in human volunteers was observed, the startle response was elicited using white-noise stimuli. In contrast, pure-tone stimuli were used in the present study. This may be of some relevance to the conclusions drawn from the lack of PPI disruption following modafinil, since a recent study found that disruptions in PPI in patients with schizophrenia were more easily detectable following the presentation of a white-noise startling stimulus compared to a pure-tone stimulus (Braff et al, 2001). It would be of interest, therefore, to compare the effects of modafinil and amphetamine on PPI in the same group of subjects using identical experimental conditions.
Presentation of the auditory pulse stimulus also resulted in an AEP, where the N1/P2 component reflects underlying cortical processing in the primary and secondary auditory cortices (Näätänen and Picton, 1987; Mauguière et al, 1995). Clonidine increased the time to the N1 peak (see Figure 3), suggesting that clonidine may slow the cortical processing of sensory information. Clonidine, similar to its effect on the EMG startle response amplitude, also reduced the amplitude on the N1/P2 component. This observation is in contrast to a previous report that, while describing the suppression of the EMG response of the ASR, failed to detect any change in the amplitude of the N1/P2 component of the AEP (Abduljawad et al, 2001). This suggests that suppression of the muscle response by clonidine is a more robust finding than suppression of the N1/P2 component of the AEP. Indeed, although a number of studies have shown that sedative drugs suppress the amplitude of the muscle response of the ASR (see above), many such drugs are without any effect on the amplitude of the N1/P2 component (clozapine: Graham et al, 2001; quetiapine: Graham et al, 2004; ketanserin: Graham et al, 2002; diphenhydramine: Curran et al, 1998; Scaife et al, 2005). On the other hand, the benzodiazepines have been reported with some consistency to be able to suppress the amplitude of the N1/P2 component (Lader, 1977; Pooviboonsuk et al, 1996; Curran et al, 1998; Abduljawad et al, 2001). The discrepancy between the effects of sedative drugs on the amplitudes of the muscle response and the N1/P2 component of the AEP suggests the involvement of different mechanisms. The mechanism in the case of the muscle response is likely to be the modulation of the tone of the orbicularis oculi muscle by the LC (see above), and this mechanism is obviously not involved in the AEP. Benzodiazepines may have an additional effect in modulating the generation of the AEP. The suppression of the amplitude of the N1/P2 component of the AEP by clonidine in the present experiment suggests that some other sedative drugs may also share this mechanism.
Interestingly, modafinil affected the N1/P2 component in an opposite direction to the effect of clonidine. Modafinil reduced the time to the P2 peak (see Figure 3), an effect that may reflect an increase in the speed of cortical processing of sensory stimuli. Although modafinil on its own had no effect on N1/P2 amplitude, it antagonized the suppressant effect of clonidine. It is tempting to speculate that the opposite effects of the two drugs reflect their opposite actions on arousal mechanisms.
Presentation of the weak prepulse before the startle-eliciting pulse produced significant PPI of the N1/P2 amplitude, in agreement with many previous reports on the N1/P2 component from our laboratory (Abduljawad et al, 1999, 2001; Phillips et al, 2000a, 2000b; Graham et al, 2001, 2002, 2004) and on various AEP components by others (Perlstein et al, 1993, 2001; Schall et al, 1996; Bender et al, 1999). None of the treatment conditions had any significant effect on PPI of the AEP (see Figure 3). The lack of effect of clonidine on PPI of the N1/P2 component is in agreement with a previous report (Abduljawad et al, 2001). It is of interest that of a number of drugs tested (reboxetine, fluvoxamine, amitriptyline: Phillips et al, 2000a; haloperidol, clozapine: Graham et al, 2001; ketanserin: Graham et al, 2002; quetiapine: Graham et al, 2004), only two (amitriptyline and clozapine) have been reported to reduce PPI of the N1/P2 complex of the AEP. The lack of effect of clonidine (α2-adrenoceptor agonist), haloperidol (D2 dopamine receptor and α1-adrenoceptor antagonist), reboxetine (noradrenaline reuptake inhibitor), ketanserin (5HT2 receptor antagonist), and fluvoxamine (5HT uptake inhibitor) argues against the involvement of noradrenergic, dopaminergic and serotonergic mechanisms. A shared feature of amitriptyline and clozapine is high affinity for muscarinic cholinoceptors, suggesting a possible role for the central cholinergic system in mediating PPI of the AEP.
The effect of the acoustic pulse stimulus could also be detected in the SCR, in agreement with previous reports (Turpin et al, 1999; Graham et al, 2005; Scaife et al, 2005). Clonidine, as well as the combination of clonidine and modafinil, reduced the amplitude of the SCR, whereas modafinil on its own had no effect on this response (see Figure 4). Because skin conductance is mediated via the sympathetic nervous system (Wallin, 1981), the reduction in the SCR by clonidine is concordant with its sympatholytic action, which in turn may reflect the ability of clonidine to ‘switch off’ LC activity (Szabadi and Bradshaw, 1996). This finding demonstrates that the reduction in the response to the startle-eliciting acoustic stimulus following the administration of clonidine can be observed not only in the EMG and AEP responses but also in the longer-latency autonomic component of the startle response. In contrast to clonidine, modafinil failed to modify the SCR, indicating that the sudomotor system is less sensitive to the sympathetic activating effect of modafinil than the pupillary and cardiovascular systems where modafinil evoked distinct effects consistent with sympathetic activation (see below). Furthermore, consistent with this finding, modafinil failed to antagonize the suppressant effect of clonidine on the SCR in contrast to its antagonistic effects on the EMG and AEP responses.
Presentation of the weak prepulse produced significant PPI of the SCR, an effect that has not previously been investigated for this component of the startle response. It is interesting that a weak prepulse can inhibit the effect of a startling stimulus on this longer-latency response in addition to the well-documented effects on the faster responses in the EMG and AEP, and this demonstrates that sensorimotor gating has widespread physiologic consequences that include influences on autonomic nervous system activity. Neither clonidine nor modafinil affected PPI of the SCR (see Figure 4). This is in agreement with the effects of clonidine and modafinil on PPI of the EMG and AEP responses (see above) and suggests that the mechanism underlying PPI of these three responses is unaffected by noradrenergic modulation by either drug.
The effect of clonidine on autonomic responses is in agreement with previous reports. Clonidine reduced pupil diameter (Bitsios et al, 1996; Phillips et al, 2000c, 2000d; Hou et al, 2005, 2006), reduced systolic and diastolic blood pressure in both the supine and the standing positions (Lal et al, 1975; Berlan et al, 1989; Morley et al, 1991; Bitsios et al, 1996; Kumari et al, 1996; Abduljawad et al, 1997, 2001; Arya et al, 1997; Phillips et al, 2000d; Hou et al, 2005, 2006), reduced salivation (Bitsios et al, 1996; Abduljawad et al, 1997, 2001; Arya et al, 1997; Phillips et al, 2000d; Hou et al, 2005, 2006), and reduced core temperature (Arya et al, 1997; Hou et al, 2006) (see Figure 5 and Table 2). The autonomic effects of clonidine are consistent with the stimulation of inhibitory α2-adrenoceptors on central neurons. The stimulation of autoreceptors on the noradrenergic cell bodies would ‘switch off’ the activity of the LC and other noradrenergic nuclei (ie A1/A5) leading to a central sympatholytic effect, manifesting as miosis, hypotension, and hypothermia, whereas the stimulation of postsynaptic receptors on salivatory neurons would result in a parasympatholytic effect, manifesting as hyposalivation (Szabadi and Bradshaw, 1996; Szabadi and Tavernor, 1999).
In contrast to the robust effects of clonidine on autonomic functions (see above), modafinil had relatively minor effects. The autonomic effects of modafinil were in opposite directions to those of clonidine, consistent with sympathetic activation, which in turn may be related to the activation of the LC (Hou et al, 2005; Taneja et al, 2005). Thus, modafinil caused a significant increase in pupil diameter at the highest luminance level, in agreement with a previous report using a 200-mg dose of modafinil (Hou et al, 2005, 2007). Modafinil also appeared to increase blood pressure, although only the change in supine systolic blood pressure reached statistical significance. This small increase is in agreement with previous reports on the effect of modafinil on blood pressure (200 mg: Makris et al, 2004; Turner et al, 2003; Samuels et al, 2006; >200 mg: Caldwell et al, 2000; Rush et al, 2002; Makris et al, 2004; Taneja et al, 2005). In addition, when clonidine and modafinil were administered together, modafinil antagonized the effect of clonidine on supine systolic blood pressure. In the present study, modafinil did not affect core temperature although previously modafinil has produced small increases in core temperature (Brun et al, 1998; Samuels et al, 2006).
The effects of clonidine and modafinil on the plasma concentrations of the hormones prolactin, TSH and GH, shown in Figure 6, were consistent with the complex catecholaminergic regulation of these pituitary hormones (see Figure 7 in Samuels et al, 2006) and showed that the single doses used by us were also ‘endocrinologically active.’ Clonidine enhanced the plasma concentration of TSH but failed to affect the concentration of the other two hormones. The enhancement of TSH level by clonidine is in agreement with a previous report on the effect of medetomidine, another α2-adrenoceptor agonist (Kallio et al, 1988). The noradrenergic system exerts a stimulatory influence on TSH secretion (see Samuels et al, 2006), partly by enhancing stimulatory thyrotropin-releasing hormone secretion via excitatory α1-adrenoceptors and partly by reducing inhibitory somatotropin secretion via inhibitory α2-adrenoceptors. The stimulatory influence of clonidine on TSH secretion is likely to reflect the stimulation of postsynaptic α2-adrenoceptors on tuberoinfundibular neurons inhibiting the secretion of somatostatin. Although clonidine caused some increase in GH secretion, this increase failed to reach statistical significance. Although the ability of clonidine to enhance GH secretion is well established (Lal et al, 1975; Lancranjan and Marbach, 1977; Gaspar et al, 1984; Murphy et al, 1984; Hunt et al, 1986), it is recognized that there are great individual differences in the GH-secretion-stimulating effect of the drug (Lal et al, 1975; Lancranjan and Marbach, 1977; Hunt et al, 1986; Vasconcellos and Spritzer, 2004). These variations in sensitivity, coupled with inter-individual variations in the time to peak of the response (Gaspar et al, 1984; Price et al, 1986; Cavallo et al, 1990), would have militated against the detection of a more robust effect in our relatively small sample. As in the case of TSH secretion, the stimulatory effect of clonidine on GH secretion is likely to be due to the attenuation of the inhibitory influence of somatostatin on the secretion of the hormone. Clonidine had no effect on the plasma concentration of prolactin, in agreement with a previous report (Lal et al, 1975). Modafinil reduced prolactin concentration, in agreement with our own previous report (Samuels et al, 2006), probably reflecting the enhancement of the noradrenergic stimulation of prolactin-release-inhibiting dopaminergic mechanisms in the arcuate nucleus. Modafinil had no effect on the plasma concentrations of TSH and GH, confirming previous observations (Brun et al, 1998; Samuels et al, 2006).

The central role of the LC in the regulation of arousal, autonomic function and motoneuron activity. Arousal: the LC sends an inhibitory output to the VLPO, a sleep-promoting area that inhibits the tuberomamillary nucleus (TMN). Both the LC and the TMN send major wakefulness-promoting excitatory outputs to the cerebral cortex to enhance alertness (Nelson et al, 2002, 2003; Hou et al, 2005). Autonomic function: the LC and the PVN of the hypothalamus are major pre-autonomic nuclei controlling sympathetic function. This is exemplified by pupillary and sweat gland activities. The LC is involved in the autonomic control of the pupil via projections to the preganglionic sympathetic neurons in the IML, which projects to the peripheral sympathetic superior cervical ganglion (SCG) that innervates the dilator muscle in the iris. The LC also inhibits the parasympathetic pathway controlling pupil diameter via a projection to the Edinger-Westphal nucleus (EW). The EW receives input from the pretectal nucleus (PT), which conveys light information from the retina, and projects to the ciliary ganglion (CG) that innervates the constrictor muscle in the iris (pupillary light reflex pathway) (Szabadi and Bradshaw, 1996). The cholinergic neurons in the sympathetic ganglia (SG) innervating sweat glands receive inputs from the IML, which in turn is under control from the PVN and LC. Motoneuron activity is facilitated by noradrenergic outputs from the LC. Both components (motor and autonomic) of the acoustic startle response are under noradrenergic modulation from the LC. The acoustic stimulus activates motoneurons in the facial nucleus (VII) and preautonomic neurons in the PVN and LC via relay neurons in the cochlea root nucleus (Crn) and caudal pontine reticular nucleus (PnC). The LC, apart from directly contributing to central sympathetic outflow, modulates facial motoneuron and PVN activity (see above) and thus the magnitude of the acoustic startle response (see text for details). Excitatory connections, solid lines; inhibitory connections, broken lines.
Although the present results per se do not allow us to infer the neuronal circuitry involved in the intimately linked regulation of arousal and startle responses, taken together with the published literature (eg Davis, 1980; De Sarro et al, 1987; Morgan et al, 1993; Sallinen et al, 1998; Hou et al, 2005; Samuels et al, 2006) they highlight the importance of the central noradrenergic system in controlling both functions. The LC may play a role in modulating both muscular and autonomic startle responses (see Figure 7). The noradrenergic facilitation of the activity of motor neurons is well documented, and attenuation of this facilitation by sedative drugs may underlie the inhibitory effect of these drugs on muscular startle responses. Furthermore, startle-eliciting acoustic stimuli lead to sympathetic activation manifesting as increases in BP (Baudrie et al, 1997; Holand et al, 1999; Girard et al, 2001) and sweat gland activity (present SCR results, see above), suggesting that the startle circuit impinges on central preautonomic neurons located in the paraventricular nucleus (PVN) of the hypothalamus and the LC, which in turn facilitate the activity of preganglionic sympathetic neurons in the intermediolateral (IML) cell column of the spinal cord. LC activation by an acoustic stimulus would be expected to be reflected in pupillary function because an increase in LC activity results in pupil dilatation and inhibition of the light reflex response (Szabadi and Bradshaw, 1996). The pupillary response to a startle-eliciting acoustic stimulus has not been studied so far, probably due to the difficulty of recording pupillary changes at the time of the eye-blink response.
In conclusion, the results of the present study have revealed several important points regarding the acoustic startle response paradigm: (i) Clonidine reduces the muscular startle response elicited in the orbicularis oculi muscle by a loud auditory stimulus, and this reduction can be antagonized by modafinil, although modafinil has no effect when administered alone, (ii) modafinil does not disrupt PPI of the muscular response, indicating that the mechanism of action of modafinil is distinct from that of amphetamine, (iii) clonidine reduces the AEP response to the auditory stimulus, whereas modafinil facilitates the AEP response (shortening of latency), indicating reduced and increased speed, respectively, of cortical processing following drug administration, (iv) clonidine reduces the SCR elicited by the auditory stimulus, and (v) significant PPI of the SCR can be observed following the presentation of a weaker prepulse before the presentation of the pulse. A comprehensive framework for interpreting the effects of sedative and alerting drugs on startle response amplitude, physiological measures of arousal and autonomic activity has been proposed to explain these results. In addition, this study has demonstrated alerting effects of a 400-mg dose of modafinil in non-sleep-deprived volunteers. The autonomic and endocrine effects following clonidine and modafinil are largely consistent with previous reports and fit with the proposed mechanisms of action of both drugs.
References
Abduljawad KAJ, Langley RW, Bradshaw CM, Szabadi E (1997). Effects of clonidine and diazepam on the acoustic startle response and on its inhibition by ‘prepulses’ in man. J Psychopharmacol 11: 29–34.
Abduljawad KAJ, Langley RW, Bradshaw CM, Szabadi E (1999). Effects of bromocriptine and haloperidol on prepulse inhibition: comparison of the acoustic startle eyeblink response and the N1/P2 auditory-evoked response in man. J Psychopharm 13: 3–9.
Abduljawad KAJ, Langley RW, Bradshaw CM, Szabadi E (2001). Effects of clonidine and diazepam on prepulse inhibition of the acoustic startle response and the N1/P2 auditory evoked potential in man. J Psychopharm 15: 237–242.
Arya DK, Langley RW, Szabadi E (1997). Comparison of the effects of high ambient temperature and clonidine on autonomic functions in man. Naunyn-Schmiedeberg's Arch Pharmacol 355: 376–383.
Baudrie V, Tulen JH, Blanc J, Elghozi JL (1997). Autonomic components of the cardiovascular responses to an acoustic startle stimulus in rats. J Auton Pharmacol 17: 303–309.
Bender S, Schall U, Wolstein J, Grzella I, Serbia D, Oades RD (1999). A topographic event-related potential follow-up study on ‘prepulse inhibition’ in first and second episode patients with schizophrenia. Psychiatry Res 90: 41–53.
Berlan M, Rascol O, Belin J, Moatti J-P, Rascol A, Montastruc J-L (1989). α2-adrenergic sensitivity in Parkinson's disease. Clin Neuropharmacol 12: 138–144.
Berridge CW, Foote SL (1991). Effects of locus coeruleus activation on electroencephalographic activity in neocortex and hippocampus. J Neurosci 11: 3135–3145.
Bitsios P, Giakoumaki SG, Frangou S (2005). The effects of dopamine agonists on prepulse inhibition in healthy men depend on baseline PPI values. Psychopharmacology (Berl) 182: 144–152.
Bitsios P, Langley RW, Szabadi E, Bradshaw CM (1996). Comparison of the effects of clonidine on tyramine- and methoxamine-evoked mydriasis in man. British J Clin Pharmacol 41: 269–275.
Bond AJ, Lader MH (1974). The use of analogue scales in rating subjective feelings. Br J Med Psychol 47: 211–218.
Bond A, Lader M, Shrotriya R (1983). Comparative effects of a repeated dose regime of diazepam and buspirone on subjective ratings, psychological tests and the EEG. Eur J Clin Pharmacol 24: 463–467.
Braff DL, Geyer MA, Light GA, Sprock J, Perry W, Cadenhead KS (2001). Impact of prepulse characteristics on the detection of sensorimotor gating deficits in schizophrenia. Schizophr Res 49: 171–178.
Brun J, Chamba G, Khalfallah Y, Girard P, Boissy I, Bastuji H (1998). Effect of modafinil on plasma melatonin, cortisol and growth hormone rhythms, rectal temperature and performance in healthy subjects during a 36 h sleep deprivation. J Sleep Res 7: 105–114.
Brunell SC, Spear LP (2006). Effects of acute ethanol or amphetamine administration on the acoustic startle response and prepulse inhibition in adolescent and adult rats. Psychopharmacology (Berl) 186: 579–586.
Bunney BS, Aghajanian GK, Roth RH (1973a). Comparison of effects of L-dopa, amphetamine and apomorphine on firing rate of rat dopaminergic neurones. Nat New Biol 245: 123–125.
Bunney BS, Walters JR, Roth RH, Aghajanian GK (1973b). Dopaminergic neurons: effect of antipsychotic drugs and amphetamine on single cell activity. J Pharmacol Exp Ther 185: 560–571.
Calcagnetti DJ, Schechter MD (1992). Psychostimulant-induced activity is attenuated by two putative dopamine release inhibitors. Pharmacol Biochem Behav 43: 1023–1031.
Caldwell Jr JA, Caldwell JL, Smythe NK, Hall KK (2000). A double-blind, placebo-controlled investigation of the efficacy of modafinil for sustaining the alertness and performance of aviators: a helicopter simulator study. Psychopharmacology (Berl) 150: 272–282.
Carasso BS, Bakshi VP, Geyer MA (1998). Disruption in prepulse inhibition after alpha-1 adrenoceptor stimulation in rats. Neuropharmacology 37: 401–404.
Cavallo A, Carskadon MA, Rosekind MR, Cattell-Harvey G (1990). Sleep, clonidine, and their interactive effect on growth hormone secretion in normal men. Psychoneuroendocrinology 15: 15–21.
Chapotot F, Pigeau R, Canini F, Bourdon L, Buguet A (2003). Distinctive effects of modafinil and D-amphetamine on the homeostatic and circadian modulation of the human waking EEG. Psychopharmacology (Berl) 166: 127–138.
Curran HV, Pooviboonsuk P, Dalton JA, Lader MH (1998). Differentiating the effects of centrally acting drugs on arousal and memory: an event-related potential study of scopolamine, lorazepam and diphenhydramine. Psychopharmacology (Berl) 135: 27–36.
Davis M (1980). Neurochemical modulation of sensory-motor reactivity: acoustic and tactile startle reflexes. Neurosci Biobehav Rev 4: 241–263.
Davis M, Gendelman DS, Tischler MD, Gendelman PM (1982). A primary acoustic startle circuit: lesion and stimulation studies. J Neurosci 2: 791–805.
Davis M, Svensson TH, Aghajanian GK (1975). Effects of d- and l-amphetamine on habituation and sensitization of the acoustic startle response in rats. Psychopharmacologia 43: 1–11.
Deroche-Gamonet V, Darnaudéry M, Bruins-Slot L, Piat F, Le Moal M, Piazza PV (2002). Study of the addictive potential of modafinil in naïve and cocaine-experienced rats. Psychopharmacology (Berl) 161: 387–395.
De Sarro GB, Ascioti C, Froio F, Libri V, Nistico G (1987). Evidence that locus coeruleus is the site where clonidine and drugs acting at alpha 1- and alpha 2-adrenoceptors affect sleep and arousal mechanisms. Br J Pharmacol 90: 675–685.
Di Chiara G, Bassareo V, fenu S, De Luca MA, Spina L, Cadoni C (2004). Dopamine and drug addiction: the nucleus accumbens shell connection. Neuropharmacology 47: 227–241.
Di Ciano P, Coury A, Depoortere RY, Egilmez Y, Lane JD, Emmett-Oglesby MW (1995). Comparison of changes in extracellular dopamine concentrations in the nucleus accumbens during intravenous self-administration of cocaine or d-amphetamine. Behav Pharmacol 6: 311–322.
Engber TM, Dennis SA, Jones BE, Miller MS, Contreras PC (1998a). Brain regional substrates for the actions of the novel wake-promoting agent modafinil in the rat: comparison with amphetamine. Neuroscience 87: 905–911.
Engber TM, Koury EJ, Dennis SA, Miller MS, Contreras PC, Bhat RV (1998b). Differential patterns of regional c-Fos induction in the rat brain by amphetamine and the novel wakefulness-promoting agent modafinil. Neurosci Lett 241: 95–98.
Ferraro L, Antonelli T, O'Connor WT, Tanganelli S, Rambert FA, Fuxe K (1997). Modafinil: an antinarcoleptic drug with a different neurochemical profile to d-amphetamine and dopamine uptake blockers. Biol Psychiatry 42: 1181–1183.
Gaspar L, Janaky T, Valkusz Z, Laszlo FA (1984). The effects of oral clonidine on the growth hormone level in acromegalic patients. J Endocrinol Invest 7: 327–329.
Girard A, Holand S, Laude D, Elghozi J-L (2001). Antihypertensive monotherapy and cardiovascular responses to an acoustic startle stimulus. J Cardiovasc Pharmacol 37: 101–107.
Graham FK (1975). The more or less startling effects of weak prestimulation. Psychophysiology 12: 238–248.
Graham SJ, Langley RW, Balboa Verduzco AM, Bradshaw CM, Szabadi E (2002). Effects of ketanserin and haloperidol on prepulse inhibition of the acoustic startle (eyeblink) response and the N1/P2 auditory evoked response in man. J Psychopharm 16: 15–22.
Graham SJ, Langley RW, Bradshaw CM, Szabadi E (2001). Effects of haloperidol and clozapine on prepulse inhibition of the acoustic startle response and the N1/P2 auditory evoked potential in man. J Psychopharm 15: 243–250.
Graham SJ, Scaife JC, Balboa VAM, Langley RW, Bradshaw CM, Szabadi E (2004). Effects of quetiapine and haloperidol on prepulse inhibition of the acoustic startle (eyeblink) response and the N1/P2 auditory evoked response in man. J Psychopharm 18: 173–180.
Graham SJ, Scaife JC, Langley RW, Bradshaw CM, Szabadi E, Xi L (2005). Effects of lorazepam on fear-potentiated startle responses in man. J Psychopharm 19: 249–258.
Holand S, Girard A, Laude D, Meyer-Bisch C, Elghozi JL (1999). Effects of an auditory startle stimulus on blood pressure and heart rate in humans. J Hypertens 17: 1893–1897.
Hou RH, Freeman C, Langley RW, Szabadi E, Bradshaw CM (2005). Does modafinil activate the locus coeruleus in man? Comparison of modafinil and clonidine on arousal and autonomic functions in human volunteers. Psychopharmacology (Berl) 181: 537–549.
Hou RH, Samuels ER, Raisi M, Langley RW, Szabadi E, Bradshaw CM (2006). Why patients with Alzheimer's disease may show increased sensitivity to tropicamide eye drops: role of locus coeruleus. Psychopharmacology (Berl) 184: 95–106.
Hou RH, Langley RW, Szabadi E, Bradshaw CM (2007). Comparison of diphenhydramine and modafinil on arousal and autonomic functions in healthy volunteers. J Psychopharm, in press (originally published online November 8, 2006, at http://www.ncbi.nlm.nih.gov).
Hunt GE, O'Sullivan BT, Johnson GF, Smythe GA (1986). Growth hormone and cortisol secretion after oral clonidine in healthy adults. Psychoneuroendocrinology 11: 317–325.
Hutchison KE, Swift R (1999). Effect of D-amphetamine on prepulse inhibition of the startle reflex in humans. Psychopharmacology (Berl) 143: 394–400.
Ivanov A, Aston-Jones G (1995). Extranuclear dendrites of locus coeruleus neurons: activation by glutamate and modulation of activity by alpha adrenoceptors. J Neurophysiol 74: 2427–2436.
Jasinski DR (2000). An evaluation of the abuse potential of modafinil using methylphenidate as a reference. J Psychopharm 14: 53–60.
Jones BE, Halaris AE, McIlhany M, Moore RY (1977). Ascending projections of the locus coeruleus in the rat. I. Axonal transport in central noradrenaline neurones. Brain Res 20: 1–21.
Jones BE, Yang T-Z (1985). The efferent projections from the reticular formation and the locus coeruleus studied by anterograde and retrograde axonal transport in the rat. J Comp Neurol 242: 56–92.
Kallio A, Koulu M, Scheinin H, Viikari J, Scheinin M (1988). Acute effects of medetomidine, a selective alpha 2-adrenoceptor agonist, on anterior pituitary hormone and cortisol secretion in man. Acta Endocrinol (Copenh) 119: 11–15.
Koch M (1999). The neurobiology of startle. Prog Neurobiol 59: 107–128.
Koch M, Schnitzler H-U (1997). The acoustic startle response in rats—circuits mediating evocation, inhibition and potentiation. Behav Brain Res 89: 35–49.
Kokkinidis L, MacNeill EP (1982). Potentiation of d-amphetamine and L-dopa-induced acoustic startle activity after long-term exposure to amphetamine. Psychopharmacology (Berl) 78: 331–335.
Kumari V, Cotter P, Corr PJ, Gray JA, Checkley SA (1996). Effect of clonidine on the human acoustic startle reflex. Psychopharmacology (Berl) 123: 353–360.
Kumari V, Mulligan OF, Cotter PA, Poon L, Toone BK, Checkley SA (1998). Effects of single oral administrations of haloperidol and d-amphetamine on prepulse inhibition of the acoustic startle reflex in healthy male volunteers. Behav Pharmacol 9: 567–576.
Lader M (1977). Effects of psychotropic drugs on auditory evoked potentials in man. In: Desmedt JE (ed). Progressive Clinical Neurophysiology. Karger: Basel. pp 142–159.
Lal S, Tolis G, Martin SB, Brown GM, Guyda H (1975). Effect of clonidine on growth hormone, prolactin, luteinizing hormone, follicle-stimulating hormone, and thyroid-stimulating hormone in the serum of normal men. J Clin Endocrinol Metab 41: 827–832.
Lancranjan I, Marbach P (1977). New evidence for growth hormone modulation by the alpha-adrenergic system in man. Metabolism 26: 1225–1230.
Lowenstein O, Feinbeig R, Lowenfeld IE (1963). Pupillary movements during acute and chronic fatigue. Invest Opthalmol 2: 138–157.
Lowenthal DT, Matzek KM, MacGregor TR (1988). Clinical pharmacokinetics of clonidine. Clin Pharmacokinetics 14: 287–310.
Lüdtke H, Wilhelm B, Adler M, Schaeffel F, Wilhelm H (1998). Mathematical procedures in data recording and processing of pupillary fatigue waves. Vision Res 38: 2889–2896.
Makris AP, Rush CR, Frederich RC, Kelly TH (2004). Wake-promoting agents with different mechanisms of action: comparison of effects of modafinil and amphetamine on food intake and cardiovascular activity. Appetite 42: 185–195.
Mansbach RS, Geyer MA, Braff DL (1988). Dopaminergic stimulation disrupts sensorimotor gating in the rat. Psychopharmacology (Berl) 94: 507–514.
Mauguière F, Cooper R, Holder GE, Luxon LM, Murray NMF (1995). Evoked potentials: normal findings by modality. In: Osselton JW (ed). Clinical Neurophysiology. Butterworth-Heinneman: Oxford. pp 364–430.
Mignot E, Nishino S, Guilleminault C, Dement WC (1994). Modafinil binds to the dopamine uptake carrier site with low affinity. Sleep 17: 436–437.
Morgan CA, Southwick SM, Grillon C, Davis M, Krystal JH, Charney DS (1993). Yohimbine-facilitated acoustic startle reflex in humans. Psychopharmacology (Berl) 110: 342–346.
Morley MJ, Bradshaw CM, Szabadi E (1991). Effects of clonidine and yohimbine on the pupillary light reflex and carbachol-evoked sweating in healthy volunteers. Br J Clin Pharmacol 31: 99–101.
Murphy MB, Brown MJ, Dollery CT (1984). Evidence for a peripheral component in the sympatholytic actions of clonidine and guanfacine in man. Eur J Clin Pharmacol 27: 23–27.
Näätänen R, Picton T (1987). The N1 wave of the human electric and magnetic response to sound: a review and an analysis of the component structure. Psychophysiology 24: 375–425.
Nelson LE, Guo TZ, Lu J, Saper CB, Franks NP, Maze M (2002). The sedative component of anesthesia is mediated by GABAA receptors in an endogenous sleep pathway. Nat Neurosci 5: 979–984.
Nelson LE, Lu J, Guo TZ, Saper CB, Franks NP, Maze M (2003). The α2-adrenoceptor agonist dexmedetomidine converges on an endogenous sleep-promoting pathway to exert its sedative effects. Anesthesiology 98: 428–436.
Nishino S, Mao J, Sampathkumaran R, Shelton J (1998). Increased dopaminergic transmission mediates the wake-promoting effects of CNS stimulants. Sleep Res Online 1: 49–61.
Norris H (1971). The action of sedatives on brain stem oculomotor systems in man. Neuropharmacology 10: 181–189.
Pack AI, Black JE, Schwartz JRL, Matheson JK (2001). Modafinil as adjunct therapy for daytime sleepiness in obstructive sleep apnea. Am J Respir Crit Care Med 164: 1675–1681.
Paladini CA, Fiorillo CD, Morikawa H, Williams JT (2001). Amphetamine selectively blocks inhibitory glutamate transmission in dopamine neurons. Nat Neurosci 4: 275–281.
Peck RE (1959). The SHP test- an aid in the detection and measurement of depression. Arch Gen Psychiatry 1: 35–40.
Perlstein WM, Fiorito E, Simons RF, Graham FK (1993). Lead stimulation effects on reflex blink, exogenous brain potentials, and loudness judgements. Psychophysiology 30: 347–358.
Perlstein WM, Simons RF, Graham FK (2001). Prepulse effects as a function of cortical projection system. Biol Psych 56: 83–111.
Phillips MA, Langley RW, Bradshaw CM, Szabadi E (2000a). The effects of some antidepressant drugs on prepulse inhibition of the acoustic startle (eyeblink) response and the N1/P2 auditory evoked response in man. J Psychopharm 14: 40–45.
Phillips MA, Oxtoby EK, Langley RW, Bradshaw CM, Szabadi E (2000b). Effects of acute tryptophan depletion on prepulse inhibition of the acoustic startle (eyeblink) response and the N1/P2 auditory evoked response in man. J Psychopharmacol 14: 258–265.
Phillips MA, Szabadi E, Bradshaw CM (2000c). Comparison of the effects of clonidine and yohimbine on pupillary diameter at different illumination levels. Br J Clin Pharmacol 50: 65–68.
Phillips MA, Szabadi E, Bradshaw CM (2000d). Comparison of the effects of clonidine and yohimbine on spontaneous pupillary fluctuations in healthy human volunteers. Psychopharmacology (Berl) 150: 85–89.
Pooviboonsuk P, Dalton JA, Curran HV, Lader MH (1996). The effects of single doses of lorazepam on event-related potentials and cognitive function. Hum Psychopharm 11: 241–252.
Price LH, Charney DS, Heninger GR (1986). Effects of trazodone treatment on alpha-2 adrenoceptor function in depressed patients. Psychopharmacology (Berl) 89: 38–44.
Rammohan KW, Rosenberg JH, Lynn DJ, Blumenfeld AM, Pollak CP, Nagaraja HN (2002). Efficacy and safety of modafinil (Provigil) for the treatment of fatigue in multiple sclerosis: a two centre phase 2 study. J Neurol Neurosurg Psychiatry 72: 179–183.
Rasmussen K, Aghajanian GK (1990). Serotonin excitation of facial motoneurons: receptor subtype characterization. Synapse 5: 324–332.
Robbins TW (1984). Cortical noradrenaline, attention and arousal. Psychol Med 14: 13–21.
Rosenthal MH, Bryant SL (2004). Benefits of adjunct modafinil in an open-label, pilot study in patients with schizophrenia. Clin Neuropharmacol 27: 38–43.
Rush CR, Kelly TH, Hays LR, Baker RW, Wooten AF (2002). Acute behavioural and physiological effects of modafinil in drug abusers. Behav Pharmacol 13: 105–115.
Sallinen J, Haapalinna A, Viitamaa T, Kobilka BK, Scheinin M (1998). Adrenergic α2C-receptors modulate the acoustic startle reflex, prepulse inhibition, and aggression in mice. J Neurosci 18: 3035–3042.
Samuels ER, Hou RH, Langley RW, Szabadi E, Bradshaw CM (2006). Comparison of pramipexole and modafinil on arousal, autonomic, and endocrine functions in healthy volunteers. J Psychopharm 20: 756–770.
Saper CB, Scammell TE (2004). Modafinil: a drug in search of a mechanism. Sleep 27: 11–12.
Saper CB, Scammell TE, Lu J (2005). Hypothalamic regulation of sleep and circadian rhythms. Nature 437: 1257–1263.
Scaife JC, Groves J, Langley RW, Bradshaw CM, Szabadi E (2006). Sensitivity of late-latency auditory and somatosensory evoked potentials to threat of electric shock and the sedative drugs diazepam and diphenhydramine in human volunteers. J Psychopharm 20: 485–495.
Scaife JC, Langley RW, Bradshaw CM, Szabadi E (2005). Diazepam suppresses the acquisition but not the expression of ‘fear-potentiation’ of the acoustic startle response in man. J Psychopharm 19: 347–356.
Schall U, Schon A, Zerbin D, Eggers C, Oades RD (1996). Event-related potentials during an auditory discrimination with prepulse inhibition in patients with schizophrenia, obsessive-compulsive disorder and healthy subjects. Int J Neurosci 84: 15–33.
Séguéla P, Watkins KC, Geffard M, Descarries L (1990). Noradrenaline axon terminals in adult rat neocortex: an immunocytochemical analysis in serial thin sections. Neuroscience 35: 249–264.
Sills TL (1999). Amphetamine dose dependently disrupts prepulse inhibition of the acoustic startle response in rats within a narrow time window. Brain Res Bull 48: 445–448.
Simon P, Hemet C, Ramassamy C, Costentin J (1995). Non-amphetaminic mechanism of stimulant locomotor effect of modafinil in mice. Eur Neuropsychopharmacol 5: 509–514.
Singewald N, Sharp T (2000). Neuroanatomical targets of anxiogenic drugs in the hindbrain as revealed by Fos immunocytochemistry. Neuroscience 98: 759–770.
Smith JM, Misiak H (1976). Critical flicker frequency (CFF) and psychotropic drugs in normal human subjects—a review. Psychopharmacology (Berl) 47: 175–182.
Swerdlow NR, Caine SB, Braff DL, Geyer MA (1992). The neural substrates of sensorimotor gating of the startle reflex: a review of recent findings and their implications. J Psychopharm 6: 176–190.
Swerdlow NR, Mansbach RS, Geyer MA, Pulvirenti L, Koob GF, Braff DL (1990). Amphetamine disruption of prepulse inhibition of acoustic startle is reversed by depletion of mesolimbic dopamine. Psychopharmacology (Berl) 100: 413–416.
Swerdlow NR, Stephany N, Wasserman LC, Talledo J, Shoemaker J, Auerbach PP (2003). Amphetamine effects on prepulse inhibition across-species: replication and parametric extension. Neuropsychopharmacology 28: 640–650.
Szabadi E, Bradshaw CM (1996). Autonomic pharmacology of α2-adrenoceptors. J Psychopharmacol (Berl) 10 (suppl 3): 6–18.
Szabadi E, Langley RW, Bradshaw CM (2002). Comparison of single doses of noradrenergic drugs on pupillary fatigue waves in a patient with excessive daytime sleepiness. J Sleep Res 11 (suppl 1): 220.
Szabadi E, Tavernor S (1999). Hypo- and hypersalivation induced by psychoactive drugs. CNS Drugs 11: 449–466.
Tanaka M, Yoshida M, Emoto H, Ishii H (2000). Noradrenaline systems in the hypothalamus, amygdala and locus coeruleus are involved in the provocation of anxiety: basic studies. Eur J Pharmacol 405: 397–406.
Taneja I, Diedrich A, Black BK, Byrne DW, Paranjape SY, Robertson D (2005). Modafinil elicits sympathomedullary activation. Hypertension 45: 612–618.
Turner DC, Robbins TW, Clark L, Aron AR, Dowson J, Sahakian BJ (2003). Cognitive enhancing effects of modafinil in healthy volunteers. Psychopharmacology (Berl) 165: 260–269.
Turpin G, Schaefer F, Boucsein W (1999). Effects of stimulus intensity, risetime, and duration on autonomic and behavioural responding: implications for the differentiation of orienting, startle, and defence responses. Psychophysiology 36: 453–463.
US Modafinil in Narcolepsy Multicentre Study Group (1998). Randomized trial of modafinil for the treatment of pathological somnolence in narcolepsy. Ann Neurol 43: 88–97.
US Modafinil in Narcolepsy Multicentre Study Group (2000). Randomized trial of modafinil as a treatment for the excessive daytime somnolence of narcolepsy. Neurology 54: 1166–1175.
VanderMaelen CP, Aghajanian GK (1980). Intracellular studies showing modulation of facial motoneurone excitability by serotonin. Nature 287: 346–347.
Vasconcellos F, Spritzer PM (2004). Increased growth hormone response to clonidine in nonobese normoinsulinemic patients with polycystic ovary syndrome. Fertil Steril 81: 108–113.
Wallin BG (1981). Sympathetic nerve activity underlying electrodermal and cardiovascular reactions in man. Psychophysiology 18: 470–476.
Walsh JK, Randazzo AC, Stone KL, Schweitzer PK (2004). Modafinil improves alertness, vigilance, and executive function during simulated night shifts. Sleep 27: 434–439.
Wesensten NJ, Belenky G, Kautz MA, Thorne DR, Reichardt RM, Balkin TJ (2002). Maintaining alertness and performance during sleep deprivation: modafinil versus caffeine. Psychopharmacology (Berl) 159: 238–247.
White SR, Fung SJ, Barnes CD (1991). Norepinephrine effects on spinal motoneurons. Prog Brain Res 88: 343–350.
Wisor JP, Eriksson KS (2005). Dopaminergic-adrenergic interactions in the wake promoting mechanism of modafinil. Neuroscience 132: 1027–1034.
Wisor JP, Nishino S, Sora I, Uhl GH, Mignot E, Edgar DM (2001). Dopaminergic role in stimulant-induced wakefulness. J Neurosci 21: 1787–1794.
Wong NY, Gorman S, McCormick GC, Grebow P (1998). Single-dose pharmacokinetics of modafinil and methylphenidate given alone or in combination in healthy male volunteers. J Clin Pharmacol 38: 276–282.
Yeomans JS, Frankland PW (1996). The acoustic startle reflex: neurons and connections. Brain Res Rev 21: 301–314.
Yoss RE, Moyer NJ, Hollenhurst RW (1970). Pupil size and spontaneous pupillary waves associated with alertness, drowsiness, and sleep. Neurology 20: 545–554.
Zhang J, Forkstam C, Engel JA, Svensson L (2000). Role of dopamine in prepulse inhibition of acoustic startle. Psychopharmacology (Berl) 149: 181–188.
Acknowledgements
ERS is an Institute of Neuroscience, University of Nottingham, Scholar.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Samuels, E., Hou, R., Langley, R. et al. Modulation of the Acoustic Startle Response by the Level of Arousal: Comparison of Clonidine and Modafinil in Healthy Volunteers. Neuropsychopharmacol 32, 2405–2421 (2007). https://doi.org/10.1038/sj.npp.1301363
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/sj.npp.1301363
Keywords
This article is cited by
-
Phasic activation of the locus coeruleus attenuates the acoustic startle response by increasing cortical arousal
Scientific Reports (2021)
-
Previous exposure to musical auditory stimulation immediately influences the cardiac autonomic responses to the postural change maneuver in women
International Archives of Medicine (2013)
-
Phasic vs Sustained Fear in Rats and Humans: Role of the Extended Amygdala in Fear vs Anxiety
Neuropsychopharmacology (2010)
-
The relationship between physiological arousal and cortical and autonomic responses to postural instability
Experimental Brain Research (2010)
-
The 5-min pupillary alertness test is sensitive to modafinil: a placebo controlled study in patients with sleep apnea
Psychopharmacology (2008)
