Thank you for visiting nature.com. You are using a browser version with limited support for CSS. To obtain
the best experience, we recommend you use a more up to date browser (or turn off compatibility mode in
Internet Explorer). In the meantime, to ensure continued support, we are displaying the site without styles
and JavaScript.
Dimension reduction helps to visualize high-dimensional datasets. These tools should be used thoughtfully and with tuned parameters. Sometimes, these methods take a second thought.
Sometimes their queer identity is one that people set apart from their science identity. Others find unique ways to integrate multiple facets of their identity.
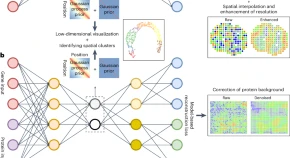
Using a dependency-aware deep generative framework, spaVAE efficiently models spatially resolved transcriptomics data and advances diverse analysis tasks. Following similar strategies, spaPeakVAE and spaMultiVAE enable spatial ATAC-seq data and spatial multi-omics data modeling and analysis, respectively.
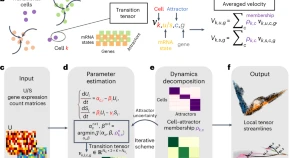
Spatial transcriptomics and mRNA splicing measurements encode rich spatiotemporal information for cell states and their transitions. We present a multiscale dynamical system method for reconstructing cell-state-specific dynamics and spatial state transitions. This theory-based approach reconciles short-timescale local tensor streamlines between cells with long-timescale transition paths that connect cell attractors.
Pebblescout navigates vast, rapidly growing nucleotide content in resources by providing indexing and search capabilities. We used Pebblescout to index a metagenomic subset of Sequence Read Archive and seven other resources into databases spanning over 3.7 petabases and searchable interactively at a pilot website using queries as short as 42 bases.
The Consortium for Top-Down Proteomics conducted a study to develop and test protocols for native mass spectrometry combined with top-down fragmentation of proteins and protein complexes across eleven instruments in nine laboratories. They report the summary of the outcomes and their recommendations in this Analysis.
OpenFold is a trainable open-source implementation of AlphaFold2. It is fast and memory efficient, and the code and training data are available under a permissive license.
By effective and efficient integration of PacBio HiFi, Oxford Nanopore Technologies ultra-long and other sequencing data types, hifiasm (UL) enables telomere-to-telomere diploid and polyploid genome assembly at a population scale.
We developed a two-pronged strategy to functionally probe the enormous repertoire of noncoding DNA within genomes. Our approach markedly improved signal-to-noise ratio and successfully intersected single-cell genomics with reporter assays. The result delivers a multiplex and highly quantitative readout of regulatory sequences’ activity in dynamic and multicellular systems.
By learning to embed DNA k-mers and cells into a joint space, CellSpace improves single-cell ATAC-seq analysis in multiple tasks such as latent structure discovery, transcription factor activity inference and batch effect mitigation.