
Abstract
We applied whole-genome resequencing of Escherichia coli to monitor the acquisition and fixation of mutations that conveyed a selective growth advantage during adaptation to a glycerol-based growth medium. We identified 13 different de novo mutations in five different E. coli strains and monitored their fixation over a 44-d period of adaptation. We obtained proof that the observed spontaneous mutations were responsible for improved fitness by creating single, double and triple site-directed mutants that had growth rates matching those of the evolved strains. The success of this new genome-scale approach indicates that real-time evolution studies will now be practical in a wide variety of contexts.
Similar content being viewed by others

Main
Comparative genomics has been almost entirely focused on genomic changes over long periods of time, on the order of millions of years. A new microarray-based method of whole-genome resequencing called comparative genome sequencing (CGS)1 now makes it cost efficient to monitor bacterial evolution comprehensively over short time periods as well. This capability is important because many microbial phenomena, such as the emergence of new pathogens and the acquisition of antibiotic resistance factors, can occur over relatively short time scales. Experimental evolution of bacteria and viruses2,3 is a facile approach to study these topics. It has been used to test predictions of evolutionary theory4 and to study parallel changes in populations evolved for 20,000 generations5, acquisition of antibiotic resistance1 and in vitro symbiosis6. Our laboratory has used experimental evolution as a tool for metabolic engineering7 and to study the recovery of strains with gene knockouts in central metabolic genes8. Nevertheless, much remains unknown about genome plasticity over short evolutionary timescales.
It is a common mistake to think of bacteria as static; that is, to assume that a culture grown overnight is the same as it was the day before. It has been estimated that nearly 10% of the individual bacteria in a Salmonella enterica population carry large-scale genome rearrangements9, and in a suboptimal environment, selection can alter a population very rapidly. The 10–20 generations that occur in the process of growing a bacterial culture are sufficient to create a heterogeneous population, depending on the magnitude of the selective advantage of adaptive mutations. This problem is avoided by using strains of bacteria that are adapted to common laboratory media, but there are interesting cases where a seemingly straightforward growth medium poses a great challenge to a bacterium.
An example is E. coli K-12 grown in minimal medium supplemented with glycerol as the carbon and energy source. Despite a complete pathway for glycerol catabolism, large variations in the growth rates of various strains have been noted10. Growth of the sequenced strain MG1655 has been observed to differ from computational predictions based on flux balance analysis11. Upon extended logarithmic growth in glycerol minimal medium, the growth rate is observed to increase from μ = 0.22 h−1 to μ = 0.55 h−1 in as few as 20 d (∼223 generations) and is observed to reach the computationally predicted optimum.
The goals of the current study were to determine the genetic basis of the adaptation to glycerol minimal medium and to observe the dynamic behavior of the mutations identified. We chose five populations of E. coli that were cultured in continuous logarithmic growth in glycerol minimal medium. Gene expression studies of these populations have been performed previously but have not uncovered the underlying basis of adaptation12. We have previously reported the results of resequencing 4.4% of the genomes of these populations using mass spectrometry13. In the current study, we have performed comprehensive resequencing of these strains, evaluated the effects of mutations both genetically and biochemically and monitored their occurrence within the populations.
Results
Comparative genome sequencing
We isolated individual clones after 44 d of growth in glycerol minimal medium (∼660 generations) from each of five populations, named GB, GC, GD, GE and G2. Genomic DNA was isolated and hybridized to microarrays by Nimblegen Systems, Inc. using their CGS strategy1. A total of 95 putative SNPs were reported by Nimblegen in the five clones. Each putative SNP was checked by PCR amplification and sequencing using MassARRAY SNP discovery (Sequenom) and reaffirmed by Sanger sequencing if detected. Out of 95 reported SNPs, 17 were confirmed (one false positive per 297 kb). Of the confirmed mutations, nine were resolved as sequence differences at three loci between the sequenced strain of MG1655 and the strain used to start the experimental evolution (see Methods).
In addition to identifying the SNPs, Nimblegen provided the location of probes that showed a difference between the test and reference samples on mapping arrays but that showed inconclusive results on the sequencing arrays. From this data, we chose 15 regions for PCR amplification and Sanger sequencing, selecting regions where multiple nearby probes were implicated. This process led to the identification of three deletions. In addition, we identified a probable duplication in clone GC-1 of a 1,313-kb region that included the replication origin between two copies of the mobile genetic element IS2 (b3044 and b4273). All confirmed mutations are listed in Table 1.
Mobile genetic elements, particularly insertion sequence (IS) elements, excise from and insert into genomic DNA at relatively high frequency. Their movement is one of the most common types of genetic changes and is considered an important factor in evolution14. The E. coli MG1655 genome contains seven different IS elements (26 total copies); the most prevalent are IS1 and IS5. To check for the movement of these IS elements, we probed DNA blots of FspI-digested genomic DNA from wild-type bacteria and from clones G1-1, G2-1, GA-1 and GD-1 with IS1 and IS5 DNA. The pattern of bands matched the pattern calculated from the reference genome sequence except for copy #5 of IS1 (b1894), which was deleted in all strains and their wild-type progenitor. The deletion was confirmed by PCR amplification using primers adjacent to IS1-5. This loss of IS1-5 is consistent with an independent resequencing project6 and high rates of activity noted previously at this locus15.
MassARRAY SNP discovery and Sanger sequencing of candidate genes representing ∼7% of the genome uncovered two additional mutations that were not detected by the microarrays, including a 9-bp duplication in glpK and a 28-bp deletion of the 3′ end of pdxK and part of the downstream transcriptional terminator in clone G2-1. It seems likely that the strong secondary structure of the terminator led to the failure to detect the pdxK deletion with CGS. A total of 1.5% of the E. coli genome consists of predicted intrinsic terminator sequences (computed using data from http://cbcb.umd.edu/software/transterm/; C. Kingsford, personal communication). Nimblegen estimates the rate of false negative detection of single nucleotide substitutions to be 5%. The error rates for the detection of deletions and insertions on the low-resolution mapping arrays is not known.
Analysis of mutations
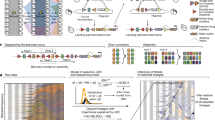
To evaluate the contributions of individual mutations to increased growth rate in glycerol minimal medium, we introduced each mutation into the wild-type strain using a site-directed mutagenesis strategy called gene gorging16. Growth rate measurements (Fig. 1) showed that mutations in genes encoding the two major subunits of RNA polymerase (rpoB and rpoC) conferred the largest change in growth rate, representing 48%–65% of the total change. Mutations in glpK and pdxK also had significant effects. The 82-bp deletion in rph probably relieves pyrimidine starvation documented in MG1655 caused by a frameshift mutation in rph17, but its effect on growth rate was undetectable. Competition experiments are a more sensitive means to measure relative fitness and may be used to investigate such mutations with small effects.

For each mutant, the names of the mutated genes are given, and in parentheses, the population from which the mutation was identified. The growth rate increase was calculated as the increase in growth rate of the mutant over the wild-type divided by the increase in growth rate of the endpoint clone over the wild-type. Error bars indicate s.d. Additional details are presented in Supplementary Table 3.
In addition to creating single mutants, we also made double and triple mutants to reconstruct the clones isolated after experimental evolution. The growth rates of four such reconstruction strains matched the growth rates of the evolved clones of which they were reconstructions (GB-1, G2-1, GE-1 and GD-1). This indicates that we were able to identify all of the important mutations in these clones and that there was not any epigenetic component to the adaptation to glycerol minimal medium. On the other hand, the growth rate of one strain did not match that of the corresponding evolved clone GC-1. Sanger sequencing of ten likely candidate genes did not uncover any other mutations. It seems probable that the 1,313-kb genomic rearrangement in this strain (which we did not attempt to reconstruct) either contributes to the growth rate or masks the presence of mutations in the duplicated region. Many of the genes implicated in this study (glpK, rpoB, rpoC and cyaA) are located in the duplicated region and may carry heterozygous mutations not detected by CGS or Sanger sequencing.
All clones had mutations in the gene for glycerol kinase (glpK), which catalyzes the first step in glycerol catabolism. This protein is subject to inhibition by fructose-1,6-bisphosphate (FBP) and unphosphorylated enzyme IIAGlc (ref. 18). Partially purified protein from cells expressing the mutant genes encoding these enzymes showed reaction rates 51%–133% higher than wild-type, and five showed reduced inhibition by FBP (Table 2). As all glycerol-adapted populations have mutations in glpK, rapid growth on glycerol can be partially attributed to altered kinetic and regulatory properties of glycerol kinase. Evidently, the rate of glycerol phosphorylation catalyzed by GlpK limits the rate of growth of wild-type E. coli MG1655 (ref. 19). The glycerol catabolic pathway generates the glycolytic intermediate dihydroxyacetone phosphate, which can be converted to methylglyoxal, a toxic metabolite when present at high concentrations10,20. It is noteworthy that we did not detect mutations in any genes relating to methylglyoxal metabolism.
We identified mutations in genes involved in peptidoglycan biosynthesis (murE and dapF) in two different clones. DapF produces the metabolite meso-2,6-diaminopimelate, whereas MurE consumes it. It is unclear what advantage these mutations confer, as site-directed mutations in these genes had negligible effects. The only exception was the glpK rpoB murE triple mutant, which grew faster than the glpK rpoB double mutant. To the best of our knowledge, there is no relationship between murE or dapF mutations and glycerol metabolism. As the mutations occurred after RNA polymerase mutations were fixed (Fig. 2), they might compensate for some detrimental side effect of RNA polymerase mutations.

The prevalence of mutations in each population was measured over the course of experimental evolution using hME genotyping and MassARRAY (Sequenom). The mutant alleles are denoted by different symbols. The average maximum error over all replicate measurements using MassEXTEND biochemistry was 0.015 and thus too small to be meaningfully represented with error bars. The lower right panel shows the growth rates of the evolving populations over time.
The gene pdxK encodes pyridoxine kinase, involved in vitamin B6 salvage. As there is no direct connection between pdxK and glycerol metabolism, it is interesting to note that pdxK is adjacent to crr, encoding enzyme IIAGlc, a regulator of glycerol kinase and a critical component in catabolite repression21. The deletion at the 3′ end of pdxK results in deletion of part of a terminator between the convergently transcribed genes pdxK and crr and might be able to attenuate enzyme IIAGlc expression through some interesting mechanism, such as antisense inhibition.
Clone G1-1 carries a mutation in cyaA, encoding another key player in catabolite repression, adenylate cyclase. The 5-bp deletion near the 3′ end of cyaA is likely to result in a truncated protein. It has been previously reported that a similarly truncated mutant of adenylate cyclase had lower activity and was not repressed by glycerol-3-phosphate22. We hypothesize that wild-type strain MG1655 grows slowly on glycerol because some critical genes are misexpressed as a result of inappropriate levels of catabolite repression and that the attenuating mutations in cyaA and pdxK/crr relieve this misexpression.
Thus, much of the increase in growth rate seems to be achieved by a transcriptional adjustment. The clones GB-1, GD-1 and GE-1 may accomplish the same effect through mutations in the genes encoding RNA polymerase. It is possible that the RNA polymerase mutations affect the level of catabolite repression by affecting RNA polymerase interactions with catabolite activator protein CAP, but this explanation seems unlikely because CAP interacts with the α subunit of RNA polymerase, whereas the mutations are in the genes encoding the β and β′ subunits23. Another hypothesis is that the RNA polymerase mutations achieve a beneficial transcriptional adjustment by affecting some other regulatory aspect, such as inhibition by guanosine tetraphosphate (ppGpp), transcriptional pausing or anti-termination. Few transcriptional commonalities were identified between the evolved populations in a previous expression study12, but that previous study looked only for commonalities present in six of the seven populations. Knowing the mutations that the populations share now allows us to analyze the expression data more productively. Looking just at data from the populations with mutations in the genes encoding RNA polymerase (GA, GB, GD, GE), we found that there were 672 ORFs with statistically significant changes in gene expression in common, using wild-type cells grown in glycerol as the reference. These ORFs had a significant overrepresentation of the following functional categories: cell motility, acid resistance and stationary phase (which were downregulated) as well as protein processing and ribosome structure (which were upregulated). Hierarchical clustering of expression data from all populations showed that GA, GB, GD and GE grouped into one clade (Supplementary Fig. 1 online).
Mutation frequencies in evolving populations
We monitored the fixation of mutations identified in endpoint clones using allele frequency estimation (AFE) (Fig. 2 and Supplementary Table 1 online). We collected samples of the evolving populations and froze them at regular intervals. Genomic DNAs from the revived samples were PCR amplified, and oligonucleotides adjacent to the mutations were extended and then quantitated using mass spectrometry, validating and calibrating each assay with artificial DNA mixtures. This technique has a limit of detection of 2% (ref. 24). In general, the dynamic behavior followed the same pattern: mutations were undetectable in early time points and then became fixed over 4–20 d. Mutations with large fitness effects (glpK or rpoBC mutations) were fixed first and more quickly relative to mutations with small effects (dapF, murE, and rph mutations), as expected. An exception was the pdxK mutation in population G2, which occurred only in the last two time points, consistent with the slow rise in growth rate of this population (Fig. 2, lower right panel).
In GB, the rpoC mutation became detectable 4 d before the glpK mutation, although both mutations became fixed simultaneously at day 10, suggesting that the addition of the glpK mutation allowed the rpoC mutants to overcome competitors. Similarly, the rpoB and glpK mutations in population GD seem to have been fixed simultaneously. The glpK and rpoC mutations in GE also seem to have been fixed simultaneously, although the glpK mutation later decreased unexpectedly. This decrease may have been due to the rise of a fitter rpoC glpK combination (as discussed in the next paragraph) that was then outcompeted when the dapF mutation occurred. Overall, AFE results are consistent with the magnitude of fitness effects observed with site-directed mutagenesis and provide detailed insight into population dynamics.
These results show the histories of the evolutionary 'winners' in the populations but do not show alternate genotypes that may have been important in the outcome of the experiment. In order to observe alternate clonal lineages and to assess the amount of variation present during the course of experimental evolution, we sequenced candidate genes in individual clones isolated from the evolving populations at day 15. For strains GB, GC, GD and GE, regions of ∼500 bp surrounding each endpoint mutation were resequenced using MassARRAY SNP discovery (Sequenom) to identify alternate alleles in important genes (Table 3). We identified four alternate glpK alleles in GD and GE but none in the others. Surprisingly, two of the alternate alleles (A218T and G217T) affected the same amino acid (Asp73) as the A218T mutation in clone GB-1. A mutation in this amino acid was identified in a previous study25 and was shown to decrease inhibition by FBP and the formation of inactive tetramers. For the A218T mutation in GD, we were able to rule out cross-contamination because the accompanying rpoC mutation from GB was not present in GD colonies. AFE analysis showed the transient occurrence of this allele in GD (Fig. 2). In GE, we observed the rpoC-C2249T allele present in the endpoint in four colonies together with a glpK allele that was not present in the endpoint clone. This suggests that the rpoC-C2249T allele occurred before the glpK alleles, in agreement with AFE results, and supports our explanation of the temporary decrease in the frequency of glpK-A113C.
Colony genotyping results were consistent with AFE results in all respects except for the frequency of the rpoB mutation in GD, which was 88% in colonies and 33% in AFE. This quantitative discrepancy can be explained by colony picking bias for large colonies, as the AFE assay was calibrated with artificial control mixtures. We measured growth rates of individual clones, but the variation in growth rates seemed to be less than the noise in growth rate measurements, making it impossible to correlate growth rates with individual genotypes.
Discussion
The genetic investigation of experimental evolution can uncover mechanisms of evolution and new connections between genotype and phenotype. Recently developed methods such as CGS now allow this investigation on a genomic scale, yielding insightful results when gene expression data may be difficult to interpret. This study has demonstrated the efficacy of genome resequencing and site-directed mutagenesis for the detection and analysis of adaptive mutations. Our results demonstrate that marked changes in phenotype can be mediated by as few as two mutations, in concordance with results in Saccharomyces cerevisiae, maize and the influenza virus26,27,28. Reconstruction of the evolved clones using mutagenesis completely accounted for the phenotypic change in four out of five cases. Additionally, we used AFE to observe the dynamic behavior of mutations as they were fixed and identified transient alleles that were lost from evolving populations.
Whole-genome resequencing is now a cost-effective tool for the study of experimental bacterial evolution. The cost of CGS resequencing services from Nimblegen Systems is currently $7,500 per clone for E. coli. This cost may be less for smaller genomes or if Nimblegen increases the density of probes on their arrays. The cost of verifying 30 putative mutations is ∼$800. The accuracy of CGS relative to other methods such as polony technology6 or sequencing by synthesis29 has not been evaluated.
CGS is designed for the identification of SNPs, but we identified deletions and a large-scale duplication as well. The mutations with the largest effects occurred in the genes encoding glycerol kinase and RNA polymerase. We did not detect any mutations fixed by genetic hitchhiking (that is, the fixation of mutations by their occurrence in the same clone as a beneficial mutation). This lack of hitchhiking is presumably due to low rates of spontaneous mutation, possibly attributable to avoiding nutrient depletion (that is, stationary phase) during adaptation. As the growth rate increased, the population that was passaged each day of experimental evolution decreased from ∼107 at the outset to ∼103 cells after 44 d. We cannot calculate mutation rates from our data because we do not know the total number of mutations that occurred—only the number that were fixed. For populations that fixed three mutations, the rate of fixation was 1.8 × 10−11 per genome per generation, taking into account the population size and number of generations on each day of evolution. If we assume 2.0 × 10−9 beneficial mutations per genome per generation30, then only about 1 out of every 100 beneficial mutations was fixed.
One of the most notable questions that this study begins to address is why wild-type E. coli K-12 is maladapted to growth on glycerol minimal medium despite a complete metabolic pathway for the substrate. Part of the explanation seems to be inappropriate levels of allosteric inhibition of glycerol kinase by FBP. This tight control is present in wild-type strains to avoid the toxic accumulation of methylglyoxal10. Another part of the explanation seems to be that the transcriptional program during growth in glycerol is not optimal and can be improved by mutations in the genes encoding RNA polymerase or in genes involved in catabolite repression. We have observed that some of the glycerol-adapted clones and site-directed mutants show growth defects in rich medium (data not shown). Evolutionary trade-offs between stress tolerance and metabolic capability have been previously associated with levels of the stationary-phase sigma factor rpoS (ref. 31). It may be that E. coli strain MG1655 has been adapted over the past 80 years by cultivation in rich medium at the expense of optimal use of glycerol.
Now that the key genes have been identified, the adaptation of E. coli to glycerol may prove to be an excellent model for the study of clonal interference and other evolutionary phenomena. The most influential mutations identified fall into two classes: those affecting a specific function (such as the rate-limiting enzyme GlpK) and those affecting global transcription patterns. Future work will show whether this reflects general principles of adaptation or if there are other important strategies for optimizing metabolism. A full appreciation of the plasticity of genomes32,33 and the capacity of bacteria to rapidly adapt to new environments will emerge as genome-scale technologies such as CGS are applied to study experimental evolution.
Methods
Experimental evolution.
A derivative of E. coli strain, K-12 MG1655 (ATCC #47076), was used for all experimental evolution cultures (see strain notes, below). The strains analyzed were generated as previously described12. Briefly, cells were grown in 250 ml M9 minimal medium + 0.2% glycerol in 500-ml Erlenmeyer flasks at 30 °C using a stir bar for aeration for 44 d (average 662 generations; see Supplementary Table 2 online). Every day, optical density (OD) measurements were made and cells were diluted into fresh medium, estimating the amount of inoculum to use such that the culture would not enter stationary phase. Samples were frozen at days 2, 4, 6, 8, 10, 15, 20, 25, 30, 35, 40 and 44.
Strain notes.
The different evolving populations were designated G1, G2, GA, GB, GC, GD and GE. In a previous study12, these populations were designated Gly1, Gly2, GlyA, etc. In another study11, the populations G1 and G2 were designated E1 and E2. Clones isolated as colonies from the endpoint populations (day 44) are designated by a clone number (such as G1-1, G2-1, GA-1, etc.). In a different study13, clones G1-1 and G2-1 were designated G1-A and G2-A. Site-directed mutants are all designated with strain names that begin with the initials BOP (Supplementary Table 3 online).
A total of seven clones from different experimentally evolved populations were subjected to CGS (G1-1, G2-1, GA-1, GB-1, GC-1, GD-1 and GE-1). Clone GA-1 was found to contain the same two mutations as clone GB-1. AFE results indicated that the two mutations in clone GB-1 were detectable in population GA from day 30 onward but did not reach complete fixation. The two mutations had nearly identical frequencies in GA, suggesting that GB may have contaminated GA between days 25 and 30. It should be noted, however, that substitutions at nucleotide 218 of glpK and deletions near nucleotide 3140 of rpoC have been repeatedly isolated in glycerol evolution experiments (A.R.J., B.Ø.P., unpublished results).
Clone G1-1 was found to have five SNPs, two deletions and a 139-kb duplication (Supplementary Table 4 online). This population was apparently started from a different strain from the others; it is wild-type for hisC, btuE and ydaC, but the others have mutations in these genes (see below). G1 was one of the first adaptive evolution experiments performed in our laboratory, and no frozen stock was saved from day 0 (the earliest saved was from day 6). In a preliminary AFE experiment, we found that the aldB, rpoS and ybhF mutations were present at 100% at day 6, supporting the notion that G1 was begun from a different starting strain. Because of uncertainty with G1-1, these results were not included in the main body of this study. The mutations present in G1-1 are listed in Supplementary Table 4, including a 1-bp deletion between rph and pyrE that was not detected by CGS (a false negative). The results of mutagenesis experiments with some of those mutations are presented in Supplementary Table 3.
The genome sequence of the strain used to inoculate the evolved populations GA, GB, GC, GD, GE and G2 differs from the genome sequence of E. coli strain MG1655 at three loci: btuE, hisC and ydaC (Supplementary Table 5 online). The evolved population G1 does not have these mutations. The original ancestral strain was MG1655 (ATCC #47076), which does not have these three mutations. Evidently, the three mutations were acquired during early handling of the strain after the G1 experiment was performed and before the other evolutions. These facts were discovered during the course of resequencing the evolved clones. During mutation validation, we noticed that some putative mutations indicated by Nimblegen (btuE, hisC and ydaC) were present in more than one clone. These mutations were confirmed in the starting strain by PCR amplification and Sanger sequencing. The CGS 'mapping' arrays detect hybridization differences between an experimental and a reference DNA. It is interesting that differences between the sequence on the chips and the reference DNA were also detected. These indications of differences were present only some of the time; the btuE deletion was indicated in 'non-called regions of interest' data for three clones and in the SNP results for one clone out of seven; the hisC mutation was indicated in SNP results for five clones and the ydaC mutation was indicated in the SNP results for three clones out of seven.
Resequencing.
Genomic DNA was prepared from endpoint clones using the DNeasy Tissue Kit (Qiagen), followed by ethanol precipitation before submission to Nimblegen Systems for CGS1. Endpoint clones GA-1 and G1-1 were resequenced first, using the m54 release of the E. coli K-12 MG1655 genome sequence (GenBank accession number NC_00096). The m56 release, which included 255 sequence corrections and the addition of 454 bp to the total genome length, was used for resequencing endpoint clones G2-1, GB-1, GD-1, GC-1 and GE-1. Putative deletions were identified from data from the first step of CGS ('mapping' arrays). Data provided by Nimblegen listed 'non-called regions of interest' (a list of probes that showed a hybridization difference between the reference and experimental samples for which no base call could be made). When two of these probes occurred within 100 bp of each other, they were flagged for further investigation. Seventeen of these flagged regions were investigated using PCR amplification and Sanger sequencing (for primers, see Supplementary Table 6 online). In cases in which a deletion was verified, the same region was amplified and sequenced in other endpoint clones, which resulted in the identification of an additional deletion not detected by the microarrays, the 1-bp deletion between rph and pyrE in G1-1 (see strain notes, above). The deletion of IS1 copy #5 was evident in 'non-called region of interest' data from endpoint clones G2-A, GB-1 and GD-1 but not in G1-1, GA-1, GC-1 and GE-1.
Mutation validation.
Oligonucleotide primer design around each putative SNP and MassARRAY SNP Discovery (Sequenom) was performed as described in ref. 13. Primers are listed in Supplementary Table 6. Briefly, the 500- to 700-bp PCR products were subjected to in vitro transcription, base-specific cleavage and analysis using MALDI-TOF mass spectrometry. The confirmed mutations were then PCR amplified and genotyped by MassARRAY hME (Sequenom) or sequenced with conventional Sanger sequencing for additional confirmation.
Mutagenesis.
Each verified mutation was introduced into the WT strain using a method called 'gene gorging'16. This method introduces any desired mutation without direct selection, and therefore leaves no antibiotic resistance gene or other trace of having been used. Details about mutagenesis and growth rate measurements can be found in Supplementary Methods online.
Biochemical measurements.
The wild-type and mutant alleles of the glpK gene were amplified by PCR and cloned into pCRT7/NT-TOPO according to the manufacturer's instructions (Invitrogen). Potential clones were checked for the presence of the correct insert in the correct orientation by restriction enzyme analysis, followed by Sanger sequencing. Protein overexpression was induced with isopropyl-β-D-thiogalactoside (IPTG). Cell-free extracts were prepared and glycerol kinase was purified using Amicon Ultra-15 size exclusion filters (Millipore), retaining the fraction between 50 kDa and 100 kDa. A sample was SDS-PAGE purified, and the 56-kDa band was cut out and analyzed with a combination of MALDI-TOF and Tandem mass spectrometry to confirm the sequence of GlpK. Enzymatic activity was measured as outlined in ref. 34. The activity of glycerol kinase was measured in the forward direction by monitoring ADP release at 340 nm by coupling pyruvate kinase and lactate dehydrogenase activity. A calibration curve was obtained using enzyme standards and chemicals purchased from Sigma Chemicals.
Allele frequency estimation by matrix-assisted laser desorption/ionization–time of flight (MALDI-TOF) mass spectrometry.
Relative frequencies of mutant and corresponding wild-type alleles at different time points of evolution were determined by analysis of MassEXTEND primer extension products and MassCLEAVE products generated from amplified genomic DNA35. Details are provided in Supplementary Methods.
Analysis of clones at day 15.
Frozen glycerol stocks from day 15 of each evolving population were struck on plates containing M9 minimal medium plus glycerol. Colonies were inoculated into liquid M9 glycerol medium and grown overnight at 30 °C. Glycerol stocks were prepared from each culture, and DNA preparations were prepared using the DNeasy Tissue kit (Qiagen). Using primers listed in Supplementary Table 6, MassARRAY SNP Discovery (Sequenom) was performed as above for SNP validation, except that only the reverse RNA strand was processed in most cases. This reduced the workload but resulted in ambiguous results for 48 out of 398 clones, almost all of which were resolved by Sanger sequencing. The PCR products that were screened for mutations were those that contained mutations in clones at the endpoint. Growth rates of individual colonies revived from frozen stocks were measured in 96 × 200 μl format in M9 minimal medium + 0.2% glycerol using a Versamax shaking plate reader (Molecular Devices) at 30 °C. Growth rates of selected colonies were also measured in 250-ml volumes of M9 glycerol medium.
Note: Supplementary information is available on the Nature Genetics website.
Accession codes
References
Albert, T.J. et al. Mutation discovery in bacterial genomes: metronidazole resistance in Helicobacter pylori. Nat. Methods 2, 951–953 (2005).
Elena, S.F. & Lenski, R.E. Evolution experiments with microorganisms: the dynamics and genetic bases of adaptation. Nat. Rev. Genet. 4, 457–469 (2003).
Wichman, H.A., Badgett, M.R., Scott, L.A., Boulianne, C.M. & Bull, J.J. Different trajectories of parallel evolution during viral adaptation. Science 285, 422–424 (1999).
Rokyta, D.R., Joyce, P., Caudle, S.B. & Wichman, H.A. An empirical test of the mutational landscape model of adaptation using a single-stranded DNA virus. Nat. Genet. 37, 441–444 (2005).
Pelosi, L. et al. Parallel changes in global protein profiles during long-term experimental evolution in Escherichia coli. Genetics 173, 1851–1869 (2006).
Shendure, J. et al. Accurate multiplex polony sequencing of an evolved bacterial genome. Science 309, 1728–1732 (2005).
Fong, S.S. et al. In silico design and adaptive evolution of Escherichia coli for production of lactic acid. Biotechnol. Bioeng. 91, 643–648 (2005).
Fong, S.S. & Palsson, B.O. Metabolic gene deletion strains of Escherichia coli evolve to computationally predicted growth phenotypes. Nat. Genet. 36, 1056–1058 (2004).
Roth, J.R. et al. Rearrangements of the bacterial chromosome: formation and applications. in Escherichia coli and Salmonella: Cellular and Molecular Biology (ed. Neidhardt, F.C.) 2256–2276 (ASM Press, Washington D.C., 1996).
Lin, E.C. Glycerol dissimilation and its regulation in bacteria. Annu. Rev. Microbiol. 30, 535–578 (1976).
Ibarra, R.U., Edwards, J.S. & Palsson, B.O. Escherichia coli K-12 undergoes adaptive evolution to achieve in silico predicted optimal growth. Nature 420, 186–189 (2002).
Fong, S.S., Joyce, A.R. & Palsson, B.O. Parallel adaptive evolution cultures of Escherichia coli lead to convergent growth phenotypes with different gene expression states. Genome Res. 15, 1365–1372 (2005).
Honisch, C., Raghunathan, A., Cantor, C.R., Palsson, B.O. & van den Boom, D. High-throughput mutation detection underlying adaptive evolution of Escherichia coli-K12. Genome Res. 14, 2495–2502 (2004).
Schneider, D. & Lenski, R.E. Dynamics of insertion sequence elements during experimental evolution of bacteria. Res. Microbiol. 155, 319–327 (2004).
Barker, C.S., Pruss, B.M. & Matsumura, P. Increased motility of Escherichia coli by insertion sequence element integration into the regulatory region of the flhD operon. J. Bacteriol. 186, 7529–7537 (2004).
Herring, C.D., Glasner, J.D. & Blattner, F.R. Gene replacement without selection: regulated suppression of amber mutations in Escherichia coli. Gene 311, 153–163 (2003).
Jensen, K.F. The Escherichia coli K-12 “wild types” W3110 and MG1655 have an rph frameshift mutation that leads to pyrimidine starvation due to low pyrE expression levels. J. Bacteriol. 175, 3401–3407 (1993).
Holtman, C.K., Pawlyk, A.C., Meadow, N.D. & Pettigrew, D.W. Reverse genetics of Escherichia coli glycerol kinase allosteric regulation and glucose control of glycerol utilization in vivo. J. Bacteriol. 183, 3336–3344 (2001).
Zwaig, N., Kistler, W.S. & Lin, E.C. Glycerol kinase, the pacemaker for the dissimilation of glycerol in Escherichia coli. J. Bacteriol. 102, 753–759 (1970).
Ferguson, G.P., Totemeyer, S., MacLean, M.J. & Booth, I.R. Methylglyoxal production in bacteria: suicide or survival? Arch. Microbiol. 170, 209–218 (1998).
Saier, M.H.J., Ramseier, T.M. & Reizer, J. Regulation of carbon utilization. in Escherichia coli and Salmonella, Vol. 1 (ed. Neidhardt, F.C.) 1325–1343 (ASM Press, Washington, D.C., 1996).
Eppler, T., Postma, P., Schutz, A., Volker, U. & Boos, W. Glycerol-3-phosphate-induced catabolite repression in Escherichia coli. J. Bacteriol. 184, 3044–3052 (2002).
Lawson, C.L. et al. Catabolite activator protein: DNA binding and transcription activation. Curr. Opin. Struct. Biol. 14, 10–20 (2004).
Ross, P., Hall, L. & Haff, L.A. Quantitative approach to single-nucleotide polymorphism analysis using MALDI-TOF mass spectrometry. Biotechniques 29, 620–629 (2000).
Liu, W.Z., Faber, R., Feese, M., Remington, S.J. & Pettigrew, D.W. Escherichia coli glycerol kinase: role of a tetramer interface in regulation by fructose 1,6-bisphosphate and phosphotransferase system regulatory protein IIIglc. Biochemistry 33, 10120–10126 (1994).
Wang, H. et al. The origin of the naked grains of maize. Nature 436, 714–719 (2005).
Zeyl, C. The number of mutations selected during adaptation in a laboratory population of Saccharomyces cerevisiae. Genetics 169, 1825–1831 (2005).
Taubenberger, J.K. et al. Characterization of the 1918 influenza virus polymerase genes. Nature 437, 889–893 (2005).
Margulies, M. et al. Genome sequencing in microfabricated high-density picolitre reactors. Nature 437, 376–380 (2005).
Gerrish, P.J. & Lenski, R.E. The fate of competing beneficial mutations in an asexual population. Genetica 102–103, 127–144 (1998).
King, T., Ishihama, A., Kori, A. & Ferenci, T. A regulatory trade-off as a source of strain variation in the species Escherichia coli. J. Bacteriol. 186, 5614–5620 (2004).
Dobrindt, U., Hentschel, U., Kaper, J.B. & Hacker, J. Genome plasticity in pathogenic and nonpathogenic enterobacteria. Curr. Top. Microbiol. Immunol. 264, 157–175 (2002).
Miller, B.G. & Raines, R.T. Identifying latent enzyme activities: substrate ambiguity within modern bacterial sugar kinases. Biochemistry 43, 6387–6392 (2004).
Janson, C.A. & Cleland, W.W. The kinetic mechanism of glycerokinase. J. Biol. Chem. 249, 2562–2566 (1974).
Bansal, A. et al. Association testing by DNA pooling: an effective initial screen. Proc. Natl. Acad. Sci. USA 99, 16871–16874 (2002).
Acknowledgements
We thank A. Fiest and S. Fong for useful discussions and M. Mosko, B. Groff and A. Maggay for technical contributions. These studies were supported by a grant from the US National Institutes of Health (NIH) (GM62791). C.D.H. was supported by NIH grant 1F32GM074590-01.
Author information
Authors and Affiliations
Contributions
C.D.H. wrote the final manuscript, compiled and analyzed all final data, identified the deletions, made the site-directed mutants and measured their growth rates. A.R. initiated, designed and led work related to data acquisition, validation, analysis and interpretation of CGS data, cloning and biochemical characterization of glpK variants, day 15 colony analysis and AFE and drafted an earlier version of the manuscript. C.H. initiated, performed, analyzed and led the work relating to mass spectrometry; conceived of using mass spectrometry to confirm CGS results and using AFE on evolving populations; and wrote portions of the manuscript. T.P. verified results from CGS, performed day 15 colony analysis and AFE and assisted with other efforts. M.K.A. measured growth rates of clones from day 15 and site-directed mutants and performed day 15 colony analysis. A.R.J. performed analysis of gene expression data.
Corresponding author
Ethics declarations
Competing interests
C.H., D.v.d.B and C.R.C. are employed by Sequenom, Inc. Technology and/or services of Sequenom, Inc. are featured in this work.
T.J.A. is employed by Nimblegen Systems, Inc. Technology and/or services of Nimblegen are featured in this work.
A provisional patent application has been filed by the University of California San Diego that includes some of the work presented in this publication.
These studies were supported by a grant from the NIH (grant GM62791). B.Ø.P. and the University of California San Diego have a financial interest in Genomatica, Inc. Although this grant has been identified for conflict of interest management based on the overall scope of the project and its potential to benefit Genomatica, Inc., the research findings included in this publication may not necessarily directly relate to the interests of Genomatica, Inc.
Supplementary information
Supplementary Fig. 1
Clustergram of gene expression data from Fong et al.12. (PDF 219 kb)
Supplementary Table 1
Allele frequency estimation results from populations at different time points of experimental evolution. (PDF 7 kb)
Supplementary Table 2
Average number of generations per day of experimental evolution. (PDF 4 kb)
Supplementary Table 3
Growth rates of site-drected mutants. (PDF 9 kb)
Supplementary Table 4
Validated mutations in clone G1-1. (PDF 4 kb)
Supplementary Table 5
Mutations in the strain used to inoculate populations GA, GB, GC, GD, GE and G2. (PDF 4 kb)
Supplementary Table 6
Oligonucleotide primers. (PDF 25 kb)
Rights and permissions
About this article
Cite this article
Herring, C., Raghunathan, A., Honisch, C. et al. Comparative genome sequencing of Escherichia coli allows observation of bacterial evolution on a laboratory timescale. Nat Genet 38, 1406–1412 (2006). https://doi.org/10.1038/ng1906
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1906
This article is cited by
-
Fast screening method to identify salinity tolerant strains of foliose Ulva species. Low salinity leads to increased organic matter of the biomass
Journal of Applied Phycology (2024)
-
Improvement of the catalytic performance of glycerol kinase from Bacillus subtilis by chromosomal site-directed mutagenesis
Biotechnology Letters (2022)
-
Mutations in adaptively evolved Escherichia coli LGE2 facilitated the cost-effective upgrading of undetoxified bio-oil to bioethanol fuel
Bioresources and Bioprocessing (2021)
-
High prevalence of multiple antibiotic resistance in clinical E. coli isolates from Bangladesh and prediction of molecular resistance determinants using WGS of an XDR isolate
Scientific Reports (2021)
-
Causal mutations from adaptive laboratory evolution are outlined by multiple scales of genome annotations and condition-specificity
BMC Genomics (2020)
