
Abstract
We identified an autosomal recessive infantile-onset symptomatic epilepsy syndrome associated with developmental stagnation and blindness. Assuming a founder effect in a large Old Order Amish pedigree, we carried out a genome-wide screen for linkage and identified a single region of homozygosity on chromosome 2p12–p11.2 spanning 5.1 cM (maximum lod score of 6.84). We sequenced genes in the region and identified a nonsense mutation in SIAT9, which is predicted to result in the premature termination of the GM3 synthase enzyme (also called lactosylceramide α-2,3 sialyltransferase). GM3 synthase is a member of the sialyltransferase family and catalyzes the initial step in the biosynthesis of most complex gangliosides from lactosylceramide. Biochemical analysis of plasma glycosphingolipids confirmed that affected individuals lack GM3 synthase activity, as marked by a complete lack of GM3 ganglioside and its biosynthetic derivatives and an increase in lactosylceramide and its alternative derivatives. Although the relationship between defects in ganglioside catabolism and a range of lysosomal storage diseases is well documented, this is the first report, to our knowledge, of a disruption of ganglioside biosynthesis associated with human disease.
Similar content being viewed by others


Main
The term 'epilepsy' is applied to a broad range of conditions characterized by the spontaneous recurrence of unprovoked seizures. The most common human epilepsies are thought to be acquired, but inheritance studies have provided evidence for a large genetic component contributing to the etiology in ∼40% of cases, with a mendelian mode of inheritance accounting for only a small proportion of these1. Although susceptibility genes have not been identified, the study of mendelian epilepsies has provided insight into the mechanisms underlying these disorders. The identification of mutations in several genes encoding ligand- or voltage-gated ion channels in familial idiopathic epilepsies has identified this particular subset of epilepsies as channelopathies. But genetic study of the mendelian symptomatic epilepsies has uncovered a more diverse etiology, involving mutations that cause disordered brain development, progressive neurodegeneration and disturbances of cortical energy metabolism.
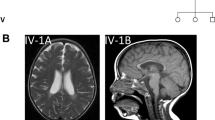
Here we identified and clinically characterized an autosomal recessive infantile-onset symptomatic epilepsy syndrome. We examined all eight surviving affected individuals from two branches of a single large pedigree (families A and B) that could be traced back to a fifth-generation common ancestor (Fig. 1). All affected children were born at full term. The first symptoms were noted between the ages of 2 weeks and 3 months, and consisted primarily of irritability with additional poor feeding, vomiting and failure to thrive. Gastrostomy feeding tubes were required in two of the four affected children in family A and all affected children in family B. Seizure activity started within the first year of life, with all affected children suffering from generalized tonic-clonic (grand mal) seizures (GTCSs), as well as other seizure types. In at least one case, startle myoclonus was observed before GTCSs. Seizure control is difficult, requiring multiple medications and, for two children in family A, vagus nerve stimulators. GTCSs were eventually largely controlled, but other seizure types including brief tonic spasms, episodes of eye deviation or startle from sleep are not fully suppressed. The onset of seizure activity is accompanied by profound developmental stagnation, with evidence of regression. Affected children are unable to sit unsupported, reach or walk and are nonverbal. Nonpurposeful arm movements have a choreoathetoid component. The children appear hypotonic with reduced or absent upper limb reflexes, although in some cases reflexes are brisk in the lower limbs. Eye contact and visual function deteriorate, probably owing to cortical impairment, although there is some evidence of optic atrophy. Electroencephalograms ultimately showed multifocal epileptiform discharges superimposed on diffuse slow background activity. Brain magnetic resonance imaging initially showed no structural abnormalities, but studies at older ages showed diffuse atrophy.

The haplotypes across the investigated interval show a region of homozygosity in all affected cases (markers in the shared region shown in red) delimited by markers D2S2180 and GATA112E03.
Genome-wide linkage analysis identified a single region of homozygosity in all eight affected children located between markers D2S2216 and D2S286 on chromosome 2. To evaluate further this 11.5-cM region, we genotyped 14 microsatellite markers in all members of the two families. All eight affected individuals were homozygous with respect to identical alleles for markers located in a 5.1-cM region between D2S2180 and GATA112E03 (Fig. 1), with a maximum multipoint lod score of 6.84 around marker D2S2162 (Fig. 2a). None of the unaffected siblings or parents were homozygous across this interval.

(a) Multipoint lod score across and beyond the region of homozygosity, flanked by markers D2S2180 and GATA112E03, producing a peak lod score of 6.84. (b) Sequence analysis of the 694C → T mutation in exon 8 of SIAT9 in samples from a wild-type control, a parent carrier and an affected individual.
The 5.1-cM region of linkage (equating to ∼6 Mb) contains ∼50 known or predicted genes. We used a high-throughput sequencing approach to screen the coding regions and associated intronic splice junctions of the genes located in the interval. We sequenced several genes (data not shown) before we identified a substitution (694C → T) in exon 8 of SIAT9 (Fig. 2b). This substitution is a nonsense mutation predicted to produce premature termination (R232X) of the protein product, GM3 synthase. Sequencing of this exon in members of families A and B showed that the substitution cosegregated perfectly with disease status: affected individuals were homozygous with respect to the mutation, parents carrying the mutation were heterozygous and unaffected siblings were either homozygous wild-type or were heterozygous carriers of the mutation. This mutation was not detected in 462 normal control chromosomes of mixed European (80%), Asian (10%) and African (10%) descent. We also screened 96 chromosomes originating from individuals of Amish descent and identified two carriers of the mutation.
GM3 synthase is a ubiquitously expressed protein present at particularly high levels in the central nervous system. The protein catalyzes the formation of GM3 ganglioside from lactosylceramide, which is the first step in the synthesis of complex ganglioside species (a- and b-series; Fig. 3). Alternative enzymatic modification of lactosylceramide leads to the synthesis of the o-series gangliosides and the globo- and neolacto-series glycosphingolipids (GSLs). The full-length GM3 synthase molecule is predicted to contain a single short transmembrane domain and two sialyl motifs, long and short, each of which possesses an invariant cysteine residue2. The nonsense mutation is located between the two sialyl motifs; therefore, the short sialyl motif is predicted not to be translated in the mutant protein product. Previous studies have shown that site-directed mutagenesis of the conserved cysteine in the short sialyl motif abolished enzymatic activity and inhibited the transportation of the sialyltransferase from the endoplasmic reticulum to the Golgi apparatus2. The nonsense mutation that we identified is therefore predicted to result in a nonfunctional protein product.

The blue box indicates all gangliosides that cannot be synthesized in the absence of GM3 synthase. Enzymes involved in the initial stages of ganglioside biosynthesis are indicated as follows: CGT, ceramide glucosyl transferase; LCS, lactosylceramide synthase; GM3S, GM3 synthase; GD3S, GD3 synthase; GalNAcT, GalNAc transferase. The main GSL species known to occur in human plasma4 are shown. The GSLs are color-coded to correspond to the species quantified in Table 1. The major ganglio- (blue; a- and b-series, brown; o-series) and alternative globo- (pink) and neolacto- (green) series GSLs derived from the immediate precursor, lactosylceramide, are delineated. The a- and b-ganglio series (blue) are absent in the plasma of the affected children that we studied. The lack of GM3, the main ganglioside in human plasma, and its biosynthetic derivatives confirms the lack of GM3 synthase activity in these individuals.
We determined the biochemical effect of this probable loss-of-function mutation in all four carrier parents, five of the affected children and four healthy unrelated controls by analyzing plasma GSL-derived oligosaccharides after fluorescently labeling them and carrying out high-performance liquid chromatography3 (HPLC; Fig. 4). We confirmed the identities of the GSL-derived oligosaccharides by comparing them with authentic standards and by sialidase digestion (data not shown). A representative profile of plasma GSLs from the normal controls showed that GM3 is the dominant ganglioside present, in agreement with a previously published study4. The neutral GSLs present in plasma included lactosylceramide, Gb3 and Gb4. The parents of affected individuals had profiles indistinguishable from those of normal controls (Fig. 4), indicating that in the liver, which is the main source of plasma derived GSLs5, the heterozygous state is compatible with normal circulating levels of GSLs. The affected children, however, completely lacked GM3 and its downstream biosynthetic derivatives but had increased levels of the immediate precursor to GM3, lactosylceramide, and evidence of increased flux through the globoside and paragloboside pathways. Quantification of the data from the analysis of ganglioside, globoside and paragloboside pathways is summarized in Table 1. The mean control plasma total ganglioside concentration of 6.4 nmol ml−1 is very similar to the values of 4.0–8.9 nmol ml−1 previously reported for human serum6.

GSLs were extracted and purified from plasma, digested with ceramide glycanase and labeled with anthranilic acid3. Neutral GSLs (pink), gangliosides (blue) and neolacto-series GSLs (green) are identified on the HPLC profiles from a representative control and family A and family B. Lactosylceramide, which elutes at 8.5 min, is not included in these profiles as its concentration is up to five times higher than that of GM3, thereby rendering peaks with longer retention times too small to distinguish in the figure. See Table 1 for plasma GSL concentrations. (The peak eluting at 24 min corresponds to the blood group antigen Lewis b and is therefore present or absent according to the genotype of the individual.)
These data on plasma GSL levels support the prediction that the 694C → T mutation in SIAT9 causes loss of function of GM3. Gangliosides are GSLs containing sialic acid that locate to the external leaflet of the plasma membrane and are particularly abundant in the central nervous system. The roles of gangliosides in the developing and adult central nervous system are not fully understood, but they are believed to function in the regulation of receptor-mediated cell-signaling pathways7,8,9,10. The biosynthesis and degradation of gangliosides are well characterized11. The complexity of GSLs results from the sequential action of glycosyltransferases in the Golgi apparatus that converts glucosylceramide into lactosylceramide and then into higher-order neutral species and gangliosides (Fig. 3). Gangliosides are either recycled by the Golgi12 or degraded in the lysosomal compartment11. Molecular defects in the degradation of GSLs leads to a range of lysosomal storage diseases, including Gaucher, Fabry, Tay-Sachs, Sandhoff and GM1 gangliosidosis13. Conversely, until now there have been no proven cases of human disorders resulting from disruption of ganglioside biosynthesis. There is precedent for the potential occurrence of such anabolic diseases. A case report from the 1970s described a child with a familial disorder characterized by increased GM3 ganglioside and N-acetylgalactosaminyltransferase deficiency, but no molecular analysis was possible at that time14.
The loss of GM3 synthase activity affects a crucial branch point in the GSL biosynthetic pathway. In the Golgi, lactosylceramide acts as the precursor for the production of several series of GSLs including the ganglio-, globo- and neolacto-series (Fig. 3). As predicted, plasma GSL analysis of individuals with mutant GM3 synthase shows a lack of flux through the GM3-catalyzed pathway, resulting in an increase in the levels of lactosylceramide and members of the alternative biosynthetic pathways. A related phenomenon was observed in a highly metastatic human melanoma cell line15, in which reduced GM3 synthase activity resulted in decreased levels of GM3, low levels of other gangliosides and a concomitant increase in lactosylceramide. We have no data on brain GSLs from affected members of these families, but o-series gangliosides may be the dominant GSL species synthesized, with the possibility of elevated lactosylceramide, depending on the relative flux through the o-series pathway in humans in the absence of GM3 synthase.
A line of GM3 synthase knockout mice has been described16. Homozygous mutant mice showed enhanced insulin sensitivity but were otherwise phenotypically normal. These mice predominantly expressed o-series gangliosides in the brain (GM1b, GD1α and GD1c), but levels of neutral GSLs were not reported. This lack of an overtly abnormal phenotype is in contrast to the severe epileptic and developmental phenotype we report in humans. The apparent disparity between the human disorder and mouse model is mirrored by observations from double knockout mice lacking both Galgt1 (encoding GM2/GD2 synthase) and Siat8a (encoding GD3 synthase). These two enzymes catalyze later stages of complex ganglioside biosynthesis by modifying GM3 (Fig. 3). Two independent double knockout lines have been established17,18. Nearly all mice from the first line have lethal audiogenic seizures17, but mice from the second line were not predisposed to audiogenic seizures18, suggesting that there might be modifier background loci in the two strains of mice19. A similar situation could account for the phenotypic differences between the epileptic human disorder described here and the milder phenotype of mice lacking GM3 synthase.
It is notable that the onset of the severe features of the disorder occurs after apparently normal intrauterine development. Although the reason for this remains unclear, a detailed analysis of GSLs in affected brain tissue or cerebrospinal fluid may help to explain this finding and elucidate the pathophysiology of this GSL biosynthetic disorder. Previous studies have reported changes in neuronal ganglioside compositions in various epileptic cohorts20,21. Of particular interest are the findings of reduced ganglioside levels in the cerebrospinal fluid of individuals with West syndrome22, a phenotypically variable infantile epileptic syndrome that has a number of similarities to the disorder reported in this study. It is not clear from our study whether the epileptic phenotype results from lack of GM3 and its derivatives or from the elevated levels of simpler GSLs caused by lack of flux through the a- and b-series ganglioside pathways. But our results provide evidence that altered GSL levels have a causal role in the etiology of this severe infantile-onset neurological disorder and implicate genes involved in ganglioside biosynthesis as candidates for causing other related human diseases.
Methods
Linkage analysis.
We extracted DNA from the blood of all 16 family members after obtaining informed consent. We carried out the initial genome-wide linkage analysis using the ABI Linkage Mapping Set Version 2.5 with an ABI 3100 sequence analyzer and Genotyper software (v3.7). We carried out marker saturation using existing and newly generated microsatellite markers. Alleles were size-fractionated using 8% polyacrylamide gels and DNA was visualized by silver staining. We generated multipoint lod scores using GENEHUNTER v2.1. We deconstructed the pedigree to facilitate computational efficiency. We assumed an autosomal recessive mode of inheritance with complete penetrance and a disease gene frequency of 0.01, estimated from the clinical incidence of the disease.
Mutation analysis.
We designed intronic primers flanking each of the nine exons of SIAT9, as well as exons of other genes located in the critical interval. We amplified these regions from 50 ng of genomic DNA using 5 pmol of each primer. We examined amplified products on 2% agarose gels and purified them for sequencing using a GENECLEAN Turbo for PCR Kit (Q-BIOgene). We sequenced PCR products using the ABI PRISM BigDye v3.1 Terminator Cycle Sequencing Ready Reaction Kit (Applied Biosystems) and analyzed them on an ABI3100 sequence analyzer. We sequenced both strands of each product.
GSL analysis.
We analyzed the release of GSL-derived oligosaccharides and carried out subsequent fluorescent labeling, separation and analysis by HPLC according to published methods3.
GenBank protein accession number.
GM3 synthase, EC 2.4.99.9.
Accession codes
References
Gardiner, R.M. Impact of our understanding of the genetic aetiology of epilepsy. J. Neurol. 247, 327–334 (2000).
Datta, A.K., Chammas, R. & Paulson, J.C. Conserved cysteines in the sialyltransferase sialylmotifs form an essential disulfide bond. J. Biol. Chem 276, 15200–15207 (2001).
Neville, D.C.A. et al. Analysis of fluorescently labelled glycosphingolipid-derived oligosaccharides following ceramide glycanase digestion and anthranilic acid labelling. Anal. Biochem. 331, 275–282 (2004).
Kundu, S.K., Diego, I., Osovitz, S. & Marcus, D.M. Glycosphingolipids of human plasma. Arch. Biochem. Biophys. 238, 388–400 (1985).
Senn, H.J. et al. Human serum gangliosides in hypercholesterolemia, before and after extracorporeal elimination of LDL. Atherosclerosis 94, 109–117 (1992).
Negroni, E., Chigorno, V., Tettamanti, G. & Sonnino, S. Evaluation of the efficiency of an assay procedure for gangliosides in human serum. Glycoconj. J. 13, 347–352 (1996).
Zeller, C.B. & Marchase, R.B. Gangliosides as modulators of cell function. Am. J. Physiol 262, C1341–C1355 (1992).
Hakomori, S. Traveling for the glycosphingolipid path. Glycoconj. J. 17, 627–647 (2000).
Kobayashi, T. & Hirabayashi, Y. Lipid membrane domains in cell surface and vacuolar systems. Glycoconj. J. 17, 163–171 (2000).
Munro, S. Lipid rafts: elusive or illusive? Cell 115, 377–388 (2003).
Sandhoff, K. & Kolter, T. Biosynthesis and degradation of mammalian glycosphingolipids. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 847–861 (2003).
Schapiro, F.B., Lingwood, C., Furuya, W. & Grinstein, S. pH-independent retrograde targeting of glycolipids to the Golgi complex. Am. J. Physiol 274, C319–C332 (1998).
Wraith, J.E. Lysosomal disorders. Semin. Neonatol. 7, 75–83 (2002).
Max, S.R. et al. GM3 (hematoside) sphingolipodystrophy. N. Engl. J. Med. 291, 929–31 (1974).
Zebda, N., Pedron, S., Rebbaa, A., Portoukalian, J. & Berthier-Vergnes, O. Deficiency of ganglioside biosynthesis in metastatic human melanoma cells: relevance of CMP-NeuAc:LacCer alpha 2-3 sialyltransferase (GM3 synthase). FEBS Lett. 362, 161–164 (1995).
Yamashita, T. et al. Enhanced insulin sensitivity in mice lacking ganglioside GM3. Proc. Natl. Acad. Sci. USA 100, 3445–3449 (2003).
Kawai, H. et al. Mice expressing only monosialoganglioside GM3 exhibit lethal audiogenic seizures. J. Biol. Chem. 276, 6885–6888 (2001).
Inoue, M. et al. Refractory skin injury in complex knock-out mice expressing only the GM3 ganglioside. J. Biol. Chem. 277, 29881–29888 (2002).
Proia, R.L. Glycosphingolipid functions: insights from engineered mouse models. Philos. Trans. R. Soc. Lond. B Biol. Sci. 358, 879–883 (2003).
Yu, R.K. & Glaser, G.H. Possible role of gangliosides in epilepsy: effects of epileptic seizures on cerebral gangliosides. Trans. Am. Neurol. Assoc. 100, 261–263 (1975).
Yu, R.K., Holley, J.A., Macala, L.J. & Spencer, D.D. Ganglioside changes associated with temporal lobe epilepsy in the human hippocampus. Yale J. Biol. Med. 60, 107–117 (1987).
Izumi, T., Ogawa, T., Koizumi, H. & Fukuyama, Y. Low levels of CSF gangliotetraose-series gangliosides in West syndrome: implication of brain maturation disturbance. Pediatr. Neurol. 9, 293–296 (1993).
Acknowledgements
We thank the Amish families for their help and support and the Das Deutsch Centre for facilitating contact with the affected individuals. This work was supported by The Birth Defects Foundation UK, The Wellcome Trust and Research to Prevent Blindness.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Rights and permissions
About this article
Cite this article
Simpson, M., Cross, H., Proukakis, C. et al. Infantile-onset symptomatic epilepsy syndrome caused by a homozygous loss-of-function mutation of GM3 synthase. Nat Genet 36, 1225–1229 (2004). https://doi.org/10.1038/ng1460
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/ng1460
This article is cited by
-
Current state-of-the-art on ganglioside-mediated immune modulation in the tumor microenvironment
Cancer and Metastasis Reviews (2023)
-
Glucosylceramide synthase inhibition reduces ganglioside GM3 accumulation, alleviates amyloid neuropathology, and stabilizes remote contextual memory in a mouse model of Alzheimer’s disease
Alzheimer's Research & Therapy (2022)
