
Abstract
The attachment of a sugar to a hydrophobic polyisoprenyl carrier is the first step for all extracellular glycosylation processes. The enzymes that perform these reactions, polyisoprenyl-glycosyltransferases (PI-GTs) include dolichol phosphate mannose synthase (DPMS), which generates the mannose donor for glycosylation in the endoplasmic reticulum. Here we report the 3.0Å resolution crystal structure of GtrB, a glucose-specific PI-GT from Synechocystis, showing a tetramer in which each protomer contributes two helices to a membrane-spanning bundle. The active site is 15 Å from the membrane, raising the question of how water-soluble and membrane-embedded substrates are brought into apposition for catalysis. A conserved juxtamembrane domain harbours disease mutations, which compromised activity in GtrB in vitro and in human DPM1 tested in zebrafish. We hypothesize a role of this domain in shielding the polyisoprenyl-phosphate for transport to the active site. Our results reveal the basis of PI-GT function, and provide a potential molecular explanation for DPM1-related disease.
Similar content being viewed by others

Introduction
In humans and bacteria alike, the first step in all glycosylation reactions that take place in outer cellular compartments involves the attachment of a sugar in the cytoplasm to a hydrophobic carrier molecule to generate a sugar donor molecule for transmembrane (TM) export1. In eukaryotes, such donors are necessary for processes including N- and O-linked protein glycosylation and glycophosphatidylinositol anchoring. In Gram-negative bacteria, they contribute to the biosynthesis of the cell wall, and its modifications that underlie serotype conversion, a bacterial mechanism for evasion of the host immune response2. Sugar attachment to hydrophobic carrier molecules for transport across membranes is catalysed by polyisoprenyl-glycosyltransferases (PI-GTs), integral membrane enzymes conserved across all kingdoms of life3. Bacterial PI-GTs utilize undecaprenyl-phosphate (UndP) as the sugar carrier, whereas their eukaryotic and archaeal counterparts use the related dolichol phosphate (DolP)3. Mutations in the human PI-GT dolichol phosphate mannose synthase (DPMS) catalytic subunit DPM1 (refs 4, 5)—the enzyme that charges DolP with mannose for protein glycosylation—cause congenital disorders of glycosylation1,6,7,8 type 1e resulting in severe phenotypes including developmental delay and vascular abnormalities9. PI-GTs have a predicted two-domain architecture composed of a cytosolic amino-terminal catalytic domain and a TM region, but their functional assemblies, detailed structures and catalytic mechanisms remain unknown.
Here we report the first structure and functional characterization of a representative PI-GT, GtrB from Synechocystis sp. PCC6803 (ref. 10; GtrBSyn) to 3.0 Å resolution. We describe enzyme assays that show that recombinant GtrBSyn expressed in Escherichia coli attaches a glucose molecule to UndP, a key reaction for immune evasion by aggressive pathogens such as Shigella flexneri and Salmonella enterica11,12. GtrB is a tetramer in which each protomer contributes two TM helices to a membrane-spanning bundle. The soluble glycosyltransferase (GT) domains are separated from the TM helices by a juxtamembrane region that positions the active site 15 Å below the membrane surface. This structure, together with results of functional mutagenesis on GtrBSyn and human DPM1 in a zebrafish model, provides the framework to understand how PI-GTs work in both prokaryotes and eukaryotes.
Results
Crystallization and structure determination
To determine the structure of a representative member of the PI-GT family, we followed a structural genomics approach, and screened 45 prokaryotic PI-GTs, identified via a bioinformatics analysis, for expression in E. coli and stability in detergents amenable to crystallization13. GtrBSyn, the PI-GT from Synechocystis sp. PCC6803, was the most promising. Indeed, we produced crystals of this protein, in the detergent n-decyl-β-D-maltopyranoside (DM), which diffracted X-rays anisotropically to 3.2/3.6 Å resolution. We determined this structure by multi-crystal single-wavelength anomalous diffraction (SAD)14 analysis of the selenomethionine-substituted protein. Subsequently, although probing the function of the enzyme with structure-based mutagenesis, we produced crystals of the point mutant F215A, which fortuitously diffracted isotropically to 3.0 Å, allowing us to improve the quality of the model (Table 1). F215A, which resides in a functionally relevant region of the protein, has compromised catalytic activity (Supplementary Table 1), but the structures are essentially identical, and unless indicated otherwise, we show here the better F215A model.
Functional characterization of GtrB from Synechocystis
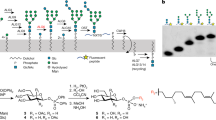
GtrB was initially identified in bacteriophage-infected Shigella flexneri strains in a cluster of genes that mediate serotype conversion by glucosylating- and acetylating-specific residues in the O-antigen of the cell wall11,15. GtrB is a bona fide PI-GT as it carries out the first step in the glucosylation process, generating UndP-glucose on the cytoplasmic face of the inner membrane (Fig. 1a), which is later utilized as a glucose donor during glucosylation of the O-antigen. To assess the PI-GT function of the enzyme from Synechocystis, we performed mass spectrometry on lipid extracts from E. coli cells overexpressing GtrBSyn, revealing specific accumulation of UndP-glucose (Fig. 1b). In vitro assays on GtrBSyn, monitoring the UndP-specific accumulation of 14C in a water-insoluble phase from UDP-14C-glucose also confirmed the synthesis of UndP-glucose from UDP-glucose and UndP (Fig. 1c).

(a) GtrB (TM domain in blue, GT domain in orange) catalyses the transfer of a glucose from UDP-glucose to UndP, producing UndP-glucose. (b) Cells overexpressing GtrB accumulate a species detected by negative ion ESI/MS at m/z of 1,007.75, corresponding to the [M-H]-ion of UndP-glucose. Its identity was further confirmed by tandem mass spectrometry (MS/MS). (c) When incubated with varying concentrations of UndP for 1 h, membranes prepared from GtrB-overexpressing cells accumulate UndP-glucose (UndP-Glc) in a concentration-dependent manner (circles), whereas those prepared from control cells overexpressing a membrane protein of unrelated function do not (squares). Error bars are provided as ±s.e.m., n=3.
The structural architecture of GtrB
The structure shows that GtrBSyn is a tetramer (Fig. 2a,b), with each protomer consisting of an N-terminal cytosolic GT domain of the GT-A fold16, a juxtamembrane region, two TM helices (TM1 and TM2), which form a compact stalk anchoring the intracellular domains to the membrane, and a short C-terminal beta-hairpin (Fig. 2c–f and Supplementary Fig. 1). The short beta-hairpin at the C-terminus bridges the interaction between adjacent subunits in the tetramer (Supplementary Fig. 2). Two amphipathic juxtamembrane helices (JM1 and JM2; Fig. 2c–f and Supplementary Fig. 1) lie at the cytoplasmic surface of the membrane, oriented parallel to one another, and sharing a hydrophobic interface (Supplementary Fig. 3). JM1 is located in an internal loop in the GT domain, whereas JM2 is an N-terminal extension of TM1 (Supplementary Fig. 1) analogous to the ‘slide helix’ found in potassium channel structures17. JM1 was first identified by analysis of the predicted secondary structure18, which suggested the presence of a conserved amphipathic helix located in the β5-6 loop and spanning residues 130–146. Although the density in this region is poorer than in other regions of the map (Supplementary Figs 4 and 5a), the presence of two methionine residues helped confirm the register of the sequence using selenomethionine-labelled protein crystals (Supplementary Fig. 5b).

The tetrameric assembly of GtrB, in which each of the four polypeptide chains is represented as a ribbon drawn in a different colour, is shown here (a) from a position orthogonal to the TM helices and (b) looking down the fourfold symmetry axis of the tetramer from the extracellular side. (c) A single protomer is shown. The protomer is comprised of a GT-A fold GT domain (orange), two amphipathic juxtamembrane helices (grey; juxtamembrane (JM)), two TM helices (blue) and a C-terminal β-hairpin (red). (d,e) The GtrB tetramer is shown in the same two views and orientations as in a,b, with three subunits represented in grey ribbons and the fourth in rainbow colouring from the N-terminus (blue) to the C-terminus (red). (f) The GtrB monomer shown in the same orientation as in c, represented in rainbow colouring from the N-terminus (N; blue) to the C-terminus (C; red).
Despite unambiguous experimental density for the majority of the molecule, electron density for the periplasmic loop that connects the TM helices is poor, leading to two possible topologies for subunit association in the tetramer (Supplementary Fig. 6). Based on our interpretation of the maps, we believe the most probable topology results in the C-terminal short beta-hairpin forming close interactions with the originating protomer (Supplementary Fig. 6a), rather than with the adjacent one (Supplementary Fig. 6b). The coordinates we have deposited leaves this ambiguity unresolved, with the protomer divided into two chains. However, for the sake of clarity, all figures showing the GtrBSyn structure depict the topology we judge to be most probable.
Donor and acceptor sites in GtrB
GtrBSyn crystals were typically soaked in UDP-glucose before cryoprotection and freezing, as this step led to better resolution and reproducibility in the quality of the diffraction pattern. This soaking step had the additional advantage of allowing us to specifically identify the monosaccharide donor site. The UDP-glucose donor substrate binds in a shallow cleft on the surface of the cytosolic GT domain, in a similar position to that determined in other structures of GT-A fold GTs19 (Fig. 3a). A divalent cation mediates the interaction between the diphosphate of the UDP and a signature DXD motif20 (94Dx96D in GtrBSyn). The anomalous signal of manganese soaked into the crystals was used to positively identify the ion-binding site although magnesium appears to be preferred for optimal activity of the enzyme (Supplementary Fig. 7a). Despite the fact that UDP-glucose was soaked into the crystals immediately before cryoprotection and freezing, interpretable density was only observed for the UDP (Supplementary Fig. 7b), indicating that the glucose is either poorly ordered or was hydrolysed before data collection, as has previously been observed for other GTs21.

(a) The disposition of key residues in the active site and interactions with the acceptor and donor substrates. An anomalous difference Fourier map (contoured at 5 σ above the mean) calculated from a tungstate-soaked wild-type (WT) crystal is represented as purple mesh. Residues D94, D96, R122, D157, R200 and the UDP are shown in stick representation. Mn2+ is represented as a purple sphere. (b) Mutation of key residues in the acceptor and donor sites abolishes GtrB activity. (c) The catalytic mechanism of GtrB does not require a catalytic base. Error bars are provided as s.e.m., n=3.
To identify the UndP acceptor site, we performed anomalous diffraction experiments with a GtrBSyn crystal soaked with tungstate, a phosphate analogue with a larger X-ray anomalous scattering signal (Fig. 3a and Supplementary Fig. 8). Remarkably, the anomalous difference density overlays perfectly with the position expected from structural alignments with other GT-acceptor complexes from soluble enzymes of the GT-A fold (Supplementary Fig. 9a).
In the 3.0 Å resolution structure without tungstate, we cannot see side chain density for R122, and the segment between R197 and N208, and the limited resolution (4.5 Å) of the data with tungstate does not allow us to reliably assign previously disordered sequence in the map.
However, we can hypothesize that R122 and R200, which are universally conserved among prokaryotic and eukaryotic GTs that process lipid-phosphate acceptors (Supplementary Fig. 9b), will coordinate the acceptor. Mutation of either of these two arginines to glutamine or alanine severely compromised GtrBSyn activity, suggesting that they play an important role in binding the phosphate of the acceptor (Fig. 3b).
The chemistry of catalysis
The transfer of a sugar group to the acceptor molecule can occur with either inversion or retention of the stereochemistry at the anomeric carbon. The corresponding two classes of GTs are known as inverting and retaining, respectively22. Overall, the data suggest that GtrB is an inverting GT, based both on previous sequence-based classification and the shallow, solvent-exposed nature of the active site seen in the GtrBSyn structure, a feature typical of inverting GTs23. The mechanism of catalysis, while broadly similar to that described for other inverting GTs, differs in detail due to the nature of the acceptor24. Unlike most GTs, which attach a saccharide donor at a neutral, protonated hydroxyl on the acceptor substrate, GtrB catalyses the attachment of glucose to the phosphate oxygen, which is expected to be deprotonated under physiological conditions. A key step in the reaction mechanism of conventional inverting GTs involves the participation of a catalytic base residue, which abstracts a proton from the acceptor, enabling nucleophilic attack by the resultant lone pair of electrons at the anomeric carbon24,25. When the acceptor is already deprotonated, it is unclear whether this step is necessary, and it seems likely that the role of a catalytic acid residue in assisting hydrolysis of the UDP-sugar donor will be more prominent. Based on structural alignment with other GT-A fold GTs and mutagenesis data showing that GtrBSyn activity is abolished in the D157N mutant (Fig. 3b), we identified D157 as a putative catalytic acid, and propose a tentative scheme for how the reaction catalysed by GtrB may take place, and describe it here in the context of the GtrBSyn structure (Fig. 3c).
Bringing substrates into apposition for catalysis to occur
The active site of GtrBSyn is located in the cytosol ∼15 Å from the membrane (Supplementary Fig. 11). This raises the question of how the hydrophobic acceptor from the membrane and hydrophilic donor from the cytosol are brought into proximity for catalysis to occur; that is, whether the GT domain moves to the membrane, or whether the lipid-phosphate acceptor is translocated to the active site. We favour the latter mechanism for three reasons. First, ∼900 Å2 of predominantly hydrophobic surface is buried between each pair of GT domains in the cytosol, with eight inter-subunit hydrogen bonds and two salt bridges formed at each interface (Supplementary Fig. 2), suggesting a stable structure that is unlikely to disassemble for the GT domain to move to the membrane. Second, the enzyme is highly specific for acceptors built from isoprenyl units (Supplementary Fig. 10), but the lack of strong conservation in the TM region (Supplementary Fig. 11) suggests that specificity for the lipid may reside elsewhere in the protein. Third, we have identified several absolutely conserved hydrophobic and basic residues in the juxtamembrane region (Supplementary Figs 11 and 12a) that could in principle form a solvent-shielded pathway for translocation of substrate from the membrane to the active site where the acceptor phosphate is bound by the side chains of R122 and R200 (Fig. 4a–c). Functional requirements for these residues (Fig. 4d), and their conserved pattern (Supplementary Figs 11 and 12a), lead us to put forth the hypothesis that as the UndP diffuses along this pathway, with its isoprenyl groups shielded by the hydrophobic residues, the phosphate headgroup could form transient ionic interactions with the positively charged moieties of the basic residues (Fig. 4a–c). To investigate this hypothesis, we first produced GtrBSyn with mutations at each of the conserved residues R122, R125, K132, A136, Y140, R200, L212, A216 and R290, in the juxtamembrane region. Mutations at any of these sites led a complete loss of enzymatic activity (Fig. 4d). However, the mutant proteins could be expressed, solubilized and purified in a non-ionic detergent at levels comparable to wild type, with a similar elution profile in size-exclusion chromatography, suggesting that the overall structure was unaffected (Supplementary Fig. 13). As further proof that the fold of GtrBSyn was unaffected by mutagenesis, we crystallized and determined the structure of several of these point mutants (Y209A, L212F, A216M, A136M, R122Q, T135A, R145A). Finally, functional assays on single-point mutations of each of the residues of JM1 and JM2 to alanine show that the entire region is extremely sensitive to even minor perturbations in structure (Supplementary Fig. 14), highlighting the importance of the juxtamembrane region in GtrB function.

(a) A speculative hypothesis is that the phosphate headgroup of UndP (red) could first bind at R290, near the cytoplasmic face of the inner membrane. (b) The substrate could then diffuse along a pathway lined with conserved hydrophobic (teal) and positively charged (blue) residues. (c) Finally, the phosphate headgroup is coordinated by R122 and R200 at the acceptor site, where catalysis occurs. (d) Mutation of hydrophobic and positively charged conserved residues lining the region between JM1 and JM2 abrogates GtrB activity. Error bars are provided as s.e.m., n=3. (e) The architecture of GtrB may be shared by the DPMS complex. CDG1e mutations G152V and S248P in the conserved JM1 and JM2 linkers are represented in purple and red, respectively. (f) Phenotypes typical of dpm1 loss-of-function mutations are fully evident at 2 days post fertilization (d.p.f.) in the zebrafish embryo. Morphants show smaller head (microcephaly) and smaller eyes, kinked tail and occasional vascular defects in the tail vein (arrowhead). Scale bar, 500 μm (g) Functional analysis of DPM1 mutants was performed by injecting human DPM1 mRNA following dpm1 morpholino injection and embryos were scored as normal or affected. Human DPM1 mRNA was able to improve the morphant phenotypes (see Supplementary Fig. 8f for details) and the effect of different mutations was normalized to rescue levels. mRNA carrying the R147A or R234A mutations (equivalent to R122 and R200 in GtrB, respectively) showed almost complete loss of function. Known human missense changes (R92S, G152V and S248P) were also tested. R92S and G152V greatly abolished protein function, whereas S248P, which leads to milder human phenotypes, showed substantial residual activity. The presumed location of G152V and S248P on the juxtamembrane region is shown. R92S is expected to reside close to the active site in the GT domain. Error bars: s.e.m., **P<0.01, ***P<0.001, unpaired Student’s t-test for each mutant, n=6 (R147A), n=6 (R234A), n=3 (R92S), n=5 (G152V), n=4 (S248P). NS, not significant.
GtrB as a structural model to study DPMS function
Although PI-GTs have high sequence variability, and they may consist of one or multiple chains, the overall architecture comprising a soluble GT domain, a juxtamembrane region composed of two amphipathic helices and one or more TM segments is conserved. The TM region of the human DPMS complex is composed of two separate polypeptide chains, but there is high conservation in the key functional residues in the juxtamembrane region we have identified here (Supplementary Fig. 12a). Interestingly, disease-causing mutations G152V and S248P in human DPM1, the catalytic subunit of the DPMS complex, have been mapped to the JM1-GT and JM2-GT linkers, and are conserved in GtrB (Fig. 4e and Supplementary Fig. 12b)26,27. Another disease mutant, R92S, which causes the most severe phenotype, is expected to be located close to the active site. Together, these observations lead us to suggest that DPMS and other PI-GTs are likely to function by similar mechanisms. To investigate this hypothesis, we performed assays to determine whether GtrB and DPMS shared functional similarities. First, we asked whether GtrB could attach its donor substrate to DolP, the acceptor used by DPMS and all other eukaryotic PI-GTs. Indeed, GtrBSyn could readily glucosylate DolP (Supplementary Fig. 12c). Second, we generated a zebrafish model of dpm1 loss of function by knocking-down dpm1 in zebrafish fertilized oocytes using two independent morpholino antisense oligonucleotides (MOs) targeting either translation (start site) or transcription (donor splice site in exon 1) at 4 and 8 ng, respectively. MO concentrations were carefully titrated to avoid nonspecific binding effects observed with high MO concentration, and knockdown was measured by quantitative PCR (Supplementary Fig. 15a,b). A scrambled control MO was used at equal concentrations to show that the effects were specific to dpm1 (Supplementary Fig. 15c). Identical phenotypes were observed for both MOs and dpm1 morphants recapitulated most features of congenital disorder of glycosylation type 1e (CDG1e) associated with DPM1 mutations, including developmental delay, microcephaly, ocular defects and vascular anomalies, which were fully evident at 2–3 days post fertilization (d.p.f.; Fig. 4f and Supplementary Fig. 15d,e)9. Injection of human DPM1 mRNA was able to significantly rescue the dpm1 morphant phenotypes showing that the function is conserved between humans and zebrafish, and that the phenotypes observed are due to the loss of dpm1 (Supplementary Fig. 15f). Injection of DPM1 mRNA alone had no effect on development (Supplementary Fig. 15g). To test whether the proposed mechanisms of action is plausible in human DPM1, we co-injected MO with mRNA carrying mutations in R147 and R234, which correspond to R122 and R200 in GtrBSyn. The mutant mRNAs failed to rescue dpm1 knockdown, suggesting that these residues are critical for function (Fig. 4g). Finally, we tested all known human mutations, R92S, G152V and S248P, which lead to variable phenotypic presentations. As observed for wild-type DPM1, overexpression of mutated mRNA alone had no effect indicating that these missense changes do not generate a gain of function (Supplementary Fig. 15g). R92S and G152V, which cause severe CDG1e, showed severe loss of function, as was previously shown in enzymatic assays9,26, whereas S248P, which causes a milder version of the disease27, only partially affected DPM1 function (Fig. 4g). Interestingly, a similar trend in activity levels was observed in GtrBSyn for the G127V (inactive) and T205P (partially inactive) mutants (Supplementary Fig. 12d).
Discussion
The structure of GtrB from Synechocystis sp. determined here at 3.0 Å resolution, is the first for a representative PI-GT, enzymes that attach a sugar to a lipid carrier for use in glycosylation reactions3. GtrBs, including GtrBSyn, attach a glucose molecule to UndP11,12, the conserved sugar-carrier lipid in prokaryotes (Fig. 1). The structure shows that GtrB is a tetramer in which each protomer has two membrane-spanning helices with a catalytic domain similar to those of soluble GTs attached through a juxtamembrane region (Fig. 2). Although the catalytic GT domain and the JM region are conserved across the family, it remains unclear whether other PI-GTs will have similar TM topologies.
Similarities in the catalytic domain with other GT-A fold GTs, coupled to functional mutagenesis data on GtrBSyn, have allowed us to identify a conserved aspartic acid (D157) as putative catalytic acid (Fig. 3b), and propose a scheme for how the reaction catalysed by GtrB may take place (Fig. 3c).
In the GtrBSyn structure, the GT domains of the tetramer are ∼15 Å distant from the membrane surface (Supplementary Fig. 11), and separated from it by the juxtamembrane region. This arrangement raises the question of how hydrophobic substrates originating from the membrane and hydrophilic substrates from the cytosol are brought into apposition for catalysis. Overall our results, including the apparent rigidity of the assembly between the four catalytic domains of the tetramer (Supplementary Fig. 2) and functional loss by mutations in the juxtamembrane region (Fig. 4d and Supplementary Fig. 14), favour a mechanism where hydrophobic polyisoprenyl-phosphates translocate from the membrane to the cytosolic-active site (Fig. 4a–c), although further studies will be required to understand the molecular details of this mechanism.
The sequence conservation we observe suggests that all PI-GTs are likely to function similarly. The GtrB structure, together with results of functional mutagenesis on this enzyme and on the human DPM1 in a zebrafish model, also presented here (Fig. 4e–g and Supplementary Fig. 14), provide a framework for understanding PI-GT structure and function in both prokaryotes and eukaryotes.
Methods
Target identification and mutagenesis
GtrB from the cyanobacterium Synechocystis sp. PCC 6803 (UniProt, accession # Q55487) was initially identified as a promising candidate for crystallization experiments by the NYCOMPS (New York Consortium on Membrane Protein Structure) high-throughput target identification and screening pipeline28,29. The open reading frame encoding GtrB was amplified by PCR using Synechocystis sp. PCC 6803 genomic DNA as template. Primers used for amplification were 5′-tacttccaatccaatgccACCATTGAACTGTCTATTGTGATT-3′ (forward) and 5′-ttatccacttccaatgCTAATTAAGTTTTTCCAATGGCAAA-3′ (reverse), where upper case letters indicate gene-specific sequences and lower case letters indicate vector-specific sequences. The amplified product was cloned into the expression vector pMCSG7 by ligation-independent cloning, introducing a hexahistidine tag and a tobacco etch virus (TEV) protease cleavage site at the N-terminus of the polypeptide30. The identity of the resulting construct was verified by DNA sequencing. Sequence alignments were performed using PRALINE and PROMALS-3D (refs 31, 32). All point mutants were prepared using the QuikChange site-directed mutagenesis kit (Agilent).
Protein expression and purification
To produce both native and the selenomethionine-substituted protein, GtrBSyn was overexpressed in E. coli strain BL21 (DE3) pLysS. Cells were grown in 2xYT medium containing 100 μg ml−1 ampicillin and 50 μg ml−1 chloramphenicol at 37 °C. At an OD600 value of 0.8, temperature was reduced to 22 °C and protein expression induced 15 min later with the addition of 0.2 mM isopropyl β-D-thiogalactopyranoside. When producing the selenomethionine-substituted protein, the protocol was the same aside from the following. Cells were grown in minimal media containing M9 salts (M9 SeMET High-Yield Growth Media Kit, Shanghai Medicilon Inc.) with a supplemental 150 mg of selenomethionine per litre of culture, and protein expression was induced with 0.4 mM isopropyl β-D-thiogalactopyranoside. The purification protocol, as described below, is the same for both native and the selenomethionine-substituted protein. Cells were harvested after overnight induction, then re-suspended in lysis buffer (20 mM Na-HEPES, pH 7.5, 150 mM NaCl, 20 mM MgSO4, 10 μg ml−1 DNase I, 10 μg ml−1 RNase A, 1 mM tris(2-carboxyethyl)phosphine (TCEP), 1 mM phenylmethanesulfonylfluoride and Complete Mini EDTA-free protease inhibitor cocktail (Roche) as described in the instructions. Cells were lysed using an Avestin Emulsiflex C3 homogenizer, and then solubilized for 1 h by addition of (Anagrade, Affymetrix) to 1% (w/v) in a typical volume of 50 ml for a pellet from an 800 ml growth culture. After pelleting insoluble debris by ultracentrifugation (134,000g, 30 min) and filtering the supernatant (0.22 μm), solubilized GtrBSyn was purified by metal-affinity followed by size-exclusion chromatography (SEC) using a 24-ml Superose 12 column (GE Healthcare). The SEC buffer contained 20 mM HEPES, pH 7.0, 150 mM NaCl, 1 mM TCEP and 0.2% (w/v) DM. Before crystallization, the peak fractions from SEC were concentrated typically to 15 mg ml−1 (estimated from A280 nm) using a centrifugal concentrator (Millipore) with a molecular weight cutoff of 50 kDa. Approximately 2 mg of purified GtrBSyn could be obtained starting from an 800-ml bacterial culture. Small-scale expression tests were performed by essentially the same protocol as for the larger-scale purification, starting from 80 mg of a cell pellet taken from a larger-scale protocol. For small-scale expression tests, cells were lysed by sonication.
Crystallization
Initial crystals were obtained by sitting drop vapour diffusion at 22 °C. The optimized crystallization protocol involved dispensing 600 nl of the purified protein solution (15 mg ml−1) and 300 nl of reservoir solution (containing 16-18% PEG600 (v/v), 0.12 M Tris/HCl, pH 9.0, 0.1 M NaCl and 1 mM TCEP) under 1 μl of silicone oil, in 96-well Axygen sitting-drop plates. Crystals typically appeared after 1–2 days and grew to a final size of ∼150 μm in each dimension. Before cryoprotection, a reservoir solution containing 5 mM MnCl2 and 25 mM UDP-glucose was added to the crystal-containing drop for 1 h. The crystals used for the tungstate anomalous difference Fourier map were soaked overnight in reservoir solution containing 10 mM sodium tungstate before cryoprotection. Crystals were flash-frozen by immersion in liquid nitrogen after incubating briefly in reservoir solution containing 28% (v/v) PEG600. Crystals of the native and the selenomethionine-substituted protein typically diffracted anisotropically to 3.2–3.8 Å resolution. GtrBSyn crystallizes in space group C2, with unit cell parameters (Å) a=157.3, b=137.5, c=101.4, β=98.5°. One tetramer is in the asymmetric unit. Crystals of the F215A mutant were produced in the same way and diffracted isotropically to 3.0 Å resolution.
Diffraction data collection and processing
Diffraction data were collected on beamline X4C at the NSLS, Brookhaven National Laboratory, and on beamline 24-ID-C at the Advanced Photon Source. Data sets collected above the Se K-edge from four selenomethionine-substituted crystals were merged and used for structure solution at 3.2 Å by multi-crystal SAD14. The data were indexed, integrated, scaled and merged using XDS and XSCALE33. Selenium sites were located using SHELXD, and refined in SHARP34. Density modification, including solvent flattening, histogram matching and four-fold non-crystallographic symmetry averaging, was performed using DM to give an initial experimentally phased electron density map35.
Model building and refinement
An initial model was built using the auto-tracing option of SHELXE, and improved using BUCCANEER36,37,38. This model was manually completed using COOT and refined using the PHENIX crystallographic software package, alternating between cycles of manual building in COOT and refinement in PHENIX39,40. Translation/Libration/Screw (TLS) parameterization was employed in the latter stages of refinement (using phenix.find_tls_groups to generate Translation/Libration/Screw (TLS) group boundaries), and riding hydrogens were included in the last cycle of refinement. Torsion-angle non-crystallographic symmetry restraints were used throughout refinement. The final models have an R/Rfree of 0.2281/0.2595 (WT) and 0.2685/0.3044 (F215A). A snapshot of typical electron density in the final 2mFo-DFc map is included as well as the anomalous difference map derived from selenomethionine-labelled wild-type crystals used for phasing and sequence registration (Supplementary Fig. 5). The quality of the final models was analysed using the validation module of the PHENIX package, which incorporates Molprobity clash score, density correlation and rotamer analysis41. For F215A, 93% of residues are in the favoured region of the Ramachandran plot, with 0.3% outliers. Full data collection and refinement statistics are given in Supplementary Table 1. Protein structure figures were prepared using UCSF Chimera42. Calculations of the surface area buried at the subunit interfaces were performed using the PISA interface in COOT43.
Mass-spectrometry analysis of lipids
The procedures for lipid extraction and liquid chromatography-mass spectrometry analysis were previously described44. Briefly, lipids were extracted from whole-cell samples (of either GtrBSyn-overexpressing cells or a negative control expressing an unrelated membrane protein) using a neutral Bligh-Dyer method45. After extraction, the dried lipids were dissolved in chloroform/methanol (2:1(v/v)). The extracted lipid species were separated by normal phase liquid chromatography (LC) using an Ascentis Silica HPLC column. LC eluents were injected into the ion spray source of a TripleTOF 5,600 quadrupole time-of-flight tandem mass spectrometer (AB SCIEX. The mass spectrometry/mass spectrometry analysis used nitrogen as the collision gas. Data analysis was performed using the Analyst TF 1.5 software.
In vitro functional assay of GtrB activity
An assay for the activity of wild-type and mutant GtrBSyn constructs using UDP-14C-glucose as a radiolabelled tracer was adapted from a previously published protocol11. Briefly, crude membranes were isolated from cell lysate by centrifugation at 134,000g for 1 h and dispersed in resuspension buffer (100 mM Tris/acetate pH 8.5) before storage at −80 °C for later use. On the day of the assay, the membranes were thawed, resuspended by sonication and washed two times with resuspension buffer. One milligram of crude membranes was used for each reaction, in a final volume of 250 μl. 1 mM EDTA, 10 mM MgCl2 and a variable (0–200 μM final concentration) amount of UndP (Indofine Chemicals) were added to the resuspended membranes, followed by a sonication step to facilitate incorporation of the UndP into the membrane. The radiolabelled substrate (0.25 μM UDP-14C-glucose) was then added and the mixture incubated at room temperature for 1 h. After extracting twice with butanol followed by two washes with water, 180 μl of the organic phase was combined with 10 volumes of scintillation fluid and radioactivity measured using a scintillation counter (Perkin Elmer). Assays testing acceptor lipids other than UndP were performed in the same way, with 100 μM of the alternate lipid in place of UndP. DolP, decaprenyl phosphate, octaprenyl phosphate and hexaprenyl phosphate were obtained from Indofine Chemicals, and dodecyl phosphate and phosphatidic acid were obtained from Sigma-Aldrich.
Alternatively, functional assays were also performed using purified protein reconstituted into liposomes. Liposomes and proteoliposomes were prepared as described46. Briefly, E. coli polar lipid extract (Avanti) and phosphatdiylcholine (PC, Avanti) were mixed in a 3:1 ratio (w/w) by dissolving in chloroform, and chloroform was removed under a stream of nitrogen gas. Lipids were resuspended in 100 mM HEPES, pH 7.5, buffer containing 1.5% (w/v) 1-O-n-Octyl-β-D-glucopyranoside (OG, Anagrade, Affymetrix) and the detergent was removed by dialysis. The resulting liposomes were divided into aliquotes, frozen in liquid nitrogen and stored at −80 °C. For proteoliposome formation, the concentration of thawed liposomes was adjusted to 10 mg ml−1 and 0.11% (w/v) Triton X-100 was added to the liposome-containing solution and purified protein was then added in a ratio of 1:80 (0.125 mg protein to 10 mg lipid), and after a 15-min incubation, detergent was removed by the addition of Bio-Beads SM-2 (Bio-Rad). Proteoliposomes were separated and removed from the Bio-Beads by careful pipetting, their concentration adjusted by ultracentrifugation and resuspension in the correct volume of buffer. Proteoliposomes were divided into aliquots, flash frozen in liquid nitrogen and stored at −80 °C. 5 μg of proteoliposomes at 10 μg ml−1 were used per assay, in a final volume of 250 μl. All subsequent steps were performed as described above for functional assays performed on membranes. Assays using protoliposomes were used to test activity of G127V and T205P GtrBSyn mutants.
Protein expression levels were normalized by quantifying the intensity of bands on an SDS–PAGE gel, after solubilization and purification by metal-affinity chromatography in DM, using ImageJ47. The expression levels of mutants were compared with wild-type protein, and activity levels were then adjusted based on this normalization.
Functional assay of human DPM1 activity in zebrafish
Fertilized zebrafish (Danio rerio) oocytes were obtained from Oregon AB breeders maintained in the Animal Research Facility at the George Washington University as described earlier48. All animal work was performed in compliance with protocols approved by the George Washington University Animal Care and Use Committees. Microinjection was performed on a PLI-100A Pico-Injector (Harvard Apparatus) at the one or two cell stage (20–45 min post fertilization) using pulled borosilicate pipettes. MO sequences were designed by Gene Tools LLC to target the start site or intron 1 splice donor site of dpm1 (ENSDART00000079591) as follows: dpm1 start 5′-TACTTCTGCGGCTCGCCATGTTGGC-3′ and dpm1 splice 5′-TTCAGGTTTCGCAATTACCTTTCGC-3′. Standard control MOs were provided by Gene Tools. MOs were injected at 1–4 ng for dpm1 start or 8–20 ng for dpm1 splice in 1 × Danieau solution (58 mM NaCl, 0.7 mM KCl, 0.4 mM MgSO4, 0.6 mM Ca(NO3)2, 5 mM HEPES, pH 7.6) with 0.1% phenol red as injection dye. Full-length or mutated DPM1 mRNA was cloned into pCS2+ for capped mRNA production. mRNA was produced using the Ambion mMessage mMachine SP6 trascription kit. Rescue injections were performed sequentially by injecting the MO and the mRNA independently at the 1–2 cell stage in 60–100 embryos per condition. For each clutch, at least four conditions were tested at the same time: un-injected wild types, MO injection, rescue (MO+wild-type mRNA), mutant (MO+mutated mRNA). Whenever sufficient embryos were available, an mRNA alone control was added or multiple mutants were tested in parallel. Embryos were maintained at 28.5 °C and staged at hours (h) and days (d) post fertilization. For imaging and analysis 2–3 d.p.f. embryos were anaesthetized using 0.4% Tricaine stock and imaged on a Leica M165 stereomicroscope. For knockdown quantification, mRNA was extracted from control and experimental embryos using Promega ReliaPrep RNA miniprep system and reverse transcribed using Bio-Rad iScript cDNA Synthesis kit. qPCR was performed using Bio-Rad SsoFast EvaGreen Supermix on a Bio-Rad CFX384 platform in the GWU Biomarker Core. Expression was normalized to ribosomal protein L8 (rpl8). Primer sequences are available in Supplementary Table 2.
Additional information
Accession codes: Coordinates and structure factors have been deposited in the Protein Data Bank under the accession code 5EKP (WT) and 5EKE (F215A).
How to cite this article: Ardiccioni, C. et al. Structure of the polyisoprenyl-phosphate glycosyltransferase GtrB and insights into the mechanism of catalysis. Nat. Commun. 7:10175 doi: 10.1038/ncomms10175 (2016).
References
Orlean, P. Dolichol phosphate mannose synthase is required in vivo for glycosyl phosphatidylinositol membrane anchoring, O mannosylation, and N glycosylation of protein in Saccharomyces cerevisiae. Mol. Cell Biol. 10, 5796–5805 (1990).
Allison, G. E. & Verma, N. K. Serotype-converting bacteriophages and O-antigen modification in Shigella flexneri. Trends Microbiol. 8, 17–23 (2000).
Bugg, T. D. & Brandish, P. E. From peptidoglycan to glycoproteins: common features of lipid-linked oligosaccharide biosynthesis. FEMS Microbiol. Lett. 119, 255–262 (1994).
Jensen, J. W. & Schutzbach, J. S. Characterization of mannosyl-transfer reactions catalyzed by dolichyl-mannosyl-phosphate-synthase. Carbohydr. Res. 149, 199–208 (1986).
Maeda, Y. & Kinoshita, T. Dolichol-phosphate mannose synthase: Structure, function and regulation. Biochim. Biophys. Acta 1780, 861–868 (2008).
Hemming, F. W. Dolichol: a curriculum cognitionis. Biochem. Cell Biol. 70, 377–381 (1992).
Imbach, T. et al. Deficiency of dolichol-phosphate-mannose synthase-1 causes congenital disorder of glycosylation type Ie. J. Clin. Invest. 105, 233–239 (2000).
Burda, P. & Aebi, M. The dolichol pathway of N-linked glycosylation. Biochim. Biophys. Acta 1426, 239–257 (1999).
Kim, S. et al. Dolichol phosphate mannose synthase (DPM1) mutations define congenital disorder of glycosylation Ie (CDG-Ie). J. Clin. Invest. 105, 191–198 (2000).
Kaneko, T. et al. Sequence analysis of the genome of the unicellular cyanobacterium Synechocystis sp. strain PCC6803. II. Sequence determination of the entire genome and assignment of potential protein-coding regions. DNA Res. 3, 109–136 (1996).
Guan, S., Bastin, D. A. & Verma, N. K. Functional analysis of the O antigen glucosylation gene cluster of Shigella flexneri bacteriophage SfX. Microbiology (Reading, Engl.) 145, 1263–1273 (1999).
Korres, H., Mavris, M., Morona, R., Manning, P. A. & Verma, N. K. Topological analysis of GtrA and GtrB proteins encoded by the serotype-converting cassette of Shigella flexneri. Biochem. Biophys. Res. Commun. 328, 1252–1260 (2005).
Mancia, F. & Love, J. High-throughput expression and purification of membrane proteins. J. Struct. Biol. 172, 85–93 (2010).
Liu, Q., Zhang, Z. & Hendrickson, W. A. Multi-crystal anomalous diffraction for low-resolution macromolecular phasing. Acta Crystallogr. D Biol. Crystallogr. 67, 45–59 (2011).
Verma, N. K., Brandt, J. M., Verma, D. J. & Lindberg, A. A. Molecular characterization of the O-acetyl transferase gene of converting bacteriophage SF6 that adds group antigen 6 to Shigella flexneri. Mol. Microbiol. 5, 71–75 (1991).
Breton, C., Snajdrová, L., Jeanneau, C., Koca, J. & Imberty, A. Structures and mechanisms of glycosyltransferases. Glycobiology 16, 29R–37R (2006).
Kuo, A. et al. Crystal structure of the potassium channel KirBac1. 1 in the closed state. Science 300, 1922–1926 (2003).
Jones, D. T. Protein secondary structure prediction based on position-specific scoring matrices. J. Mol. Biol. 292, 195–202 (1999).
Tarbouriech, N., Charnock, S. J. & Davies, G. J. Three-dimensional structures of the Mn and Mg dTDP complexes of the family GT-2 glycosyltransferase SpsA: a comparison with related NDP-sugar glycosyltransferases. J. Mol. Biol. 314, 655–661 (2001).
Wiggins, C. A. & Munro, S. Activity of the yeast MNN1 alpha-1,3-mannosyltransferase requires a motif conserved in many other families of glycosyltransferases. Proc. Natl Acad. Sci. USA 95, 7945–7950 (1998).
Larivière, L., Gueguen-Chaignon, V. & Moréra, S. Crystal structures of the T4 phage β-glucosyltransferase and the D100A mutant in complex with UDP-glucose: glucose binding and identification of the catalytic base for a direct displacement mechanism. J. Mol. Biol. 330, 1077–1086 (2003).
Kapitonov, D. & Yu, R. K. Conserved domains of glycosyltransferases. Glycobiology 9, 961–978 (1999).
Cantarel, B. L. et al. The Carbohydrate-Active EnZymes database (CAZy): an expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238 (2009).
Breton, C., Fournel-Gigleux, S. & Palcic, M. M. Recent structures, evolution and mechanisms of glycosyltransferases. Curr. Opin. Struct. Biol. 22, 540–549 (2012).
Tvaroska, I., André, I. & Carver, J. P. Ab Initio molecular orbital study of the catalytic mechanism of glycosyltransferases: description of reaction pathways and determination of transition-state structures for inverting N-acetylglucosaminyltransferases. J. Am. Chem. Soc. 122, 8762–8776 (2000).
Yang, A. C. et al. Congenital disorder of glycosylation due to DPM1 mutations presenting with dystroglycanopathy-type congenital muscular dystrophy. Mol. Genet. Metab. 110, 345–351 (2013).
García-Silva, M. T. et al. Congenital disorder of glycosylation (CDG) type Ie. A new patient. J. Inherit. Metab. Dis. 27, 591–600 (2004).
Mancia, F. & Love, J. High throughput platforms for structural genomics of integral membrane proteins. Curr. Opin. Struct. Biol. 21, 517–522 (2011).
Bruni, R. & Kloss, B. High-Throughput Cloning and Expression of Integral Membrane Proteins in Escherichia coli 29.6.1–29.6.34John Wiley & Sons, Inc. (2001).
Eschenfeldt, W. H., Lucy, S., Millard, C. S., Joachimiak, A. & Mark, I. D. A family of LIC vectors for high-throughput cloning and purification of proteins. Methods Mol. Biol. 498, 105–115 (2009).
Simossis, V. A. & Heringa, J. PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 33, W289–W294 (2005).
Pei, J., Kim, B. H. & Grishin, N. V. PROMALS3D: a tool for multiple protein sequence and structure alignments. Nucleic Acids Res. 36, 2295–2300 (2008).
Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 (2010).
Vonrhein, C., Blanc, E., Roversi, P. & Bricogne, G. Automated structure solution with autoSHARP. Methods Mol. Biol. 364, 215–230 (2007).
Cowtan, K. & Main, P. Miscellaneous algorithms for density modification. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 54, 487–493 (1998).
Sheldrick, G. M. Experimental phasing with SHELXC/D/E: combining chain tracing with density modification. Acta Crystallogr. D Biol. Crystallogr. 66, 479–485 (2010).
Cowtan, K. The Buccaneer software for automated model building. 1. Tracing protein chains. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 62, 1002–1011 (2006).
Winn, M. D. et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011).
Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010).
Adams, P. D. et al. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 (2010).
Chen, V. B. et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 66, 12–21 (2010).
Pettersen, E. F. et al. UCSF Chimera--a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612 (2004).
Krissinel, E. & Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol. 372, 774–797 (2007).
Tan, B. K. et al. Discovery of a cardiolipin synthase utilizing phosphatidylethanolamine and phosphatidylglycerol as substrates. Proc. Natl Acad. Sci. USA 109, 16504–16509 (2012).
Bligh, E. & Dyer, W. J. A rapid method of total lipid extraction and purification. Can. J. Biochem. Physiol. 37, 911–917 (1959).
Clarke, O. B. et al. Structural basis for phosphatidylinositol-phosphatebiosynthesis. Nat. Commun. 6, 9505 (2015).
Schneider, C. A., Rasband, W. S. & Eliceiri, K. W. NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012).
Lawrence, C. Advances in zebrafish husbandry and management. Methods Cell Biol. 104, 429–451 (2011).
Acknowledgements
Crystallographic data for this study were measured at beamlines X4A and X4C, of the National Synchrotron Light Source and the NE-CAT beamlines 24ID-C and E (supported by NIH-NIGMS grant P41 GM103403) at the Advanced Photon Source. This work was supported by an NIH-NIGMS initiative to the New York Consortium on Membrane Protein Structure (NYCOMPS; U54 GM095315) and by NIH-NIGMS grant R01 GM111980 to F.M., and Research Grants from the Muscular Dystrophy Association and the March of Dimes to M.C.M., C.A. was supported by a Fondazione Marche/ISSNAF Post-Doctoral Fellowship. O.B.C. was supported by a Charles H. Revson Senior fellowship. We thank Wayne A. Hendrickson for his leadership of NYCOMPS and both he and Alexander Sobolevsky for useful comments and suggestions, Naresh Verma for helpful advice, Leora Hamberger for her assistance running the Mancia Lab, and Adriana Nemes, Richard Axel and the Axel laboratory for generously providing help with functional experiments.
Author information
Authors and Affiliations
Contributions
E.K. and B.R. carried out the initial bioinformatics analysis. B.K. and R.B. did the cloning and small-scale expression tests. C.A. purified the protein and crystallized it. C.A., O.B.C. and Q.L. determined the structure. S.B. and K.R.R. assisted with data collection and processing and structure determination. Z.G. and C.L. performed the mass spectrometry experiments. O.B.C. refined the structure. C.A., O.B.C., D.T. and F.M. planned the functional experiment. C.A. and D.T. performed the functional experiments. M.C.M. planned the experiments in zebrafish. H.A.I., D.C.v.A., H.L.P. and M.C.M. carried out the experiments in zebrafish. C.A., O.B.C., M.C.M., L.S. and F.M. analysed the data and wrote the manuscript. F.M. coordinated the project.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing financial interests.
Supplementary information
Supplementary Information
Supplementary Figures 1-15, Supplementary Tables 1-2 and Supplementary References (PDF 3181 kb)
Rights and permissions
This work is licensed under a Creative Commons Attribution 4.0 International License. The images or other third party material in this article are included in the article’s Creative Commons license, unless indicated otherwise in the credit line; if the material is not included under the Creative Commons license, users will need to obtain permission from the license holder to reproduce the material. To view a copy of this license, visit http://creativecommons.org/licenses/by/4.0/
About this article

Cite this article
Ardiccioni, C., Clarke, O., Tomasek, D. et al. Structure of the polyisoprenyl-phosphate glycosyltransferase GtrB and insights into the mechanism of catalysis. Nat Commun 7, 10175 (2016). https://doi.org/10.1038/ncomms10175
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/ncomms10175
This article is cited by
-
Proteome-wide subtractive approach to prioritize a hypothetical protein of XDR-Mycobacterium tuberculosis as potential drug target
Genes & Genomics (2019)
-
Membrane association of monotopic phosphoglycosyl transferase underpins function
Nature Chemical Biology (2018)
-
Structural basis for dolichylphosphate mannose biosynthesis
Nature Communications (2017)
-
Molecular Cloning, Expression and Characterization of Oenococcus oeni Priming Glycosyltransferases
Molecular Biotechnology (2017)
-
Dolichol phosphate mannose synthase: a Glycosyltransferase with Unity in molecular diversities
Glycoconjugate Journal (2017)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
