
Abstract
Haploid Saccharomyces cerevisiae yeast cells use a prototypic cell signalling system to transmit information about the extracellular concentration of mating pheromone secreted by potential mating partners. The ability of cells to respond distinguishably to different pheromone concentrations depends on how much information about pheromone concentration the system can transmit. Here we show that the mitogen-activated protein kinase Fus3 mediates fast-acting negative feedback that adjusts the dose response of the downstream system response to match the dose response of receptor-ligand binding. This ‘dose–response alignment’, defined by a linear relationship between receptor occupancy and downstream response, can improve the fidelity of information transmission by making downstream responses corresponding to different receptor occupancies more distinguishable and reducing amplification of stochastic noise during signal transmission. We also show that one target of the feedback is a previously uncharacterized signal-promoting function of the regulator of G-protein signalling protein Sst2. Our work suggests that negative feedback is a general mechanism used in signalling systems to align dose responses and thereby increase the fidelity of information transmission.
This is a preview of subscription content, access via your institution
Access options
Subscribe to this journal
Receive 51 print issues and online access
$199.00 per year
only $3.90 per issue
Buy this article
- Purchase on Springer Link
- Instant access to full article PDF
Prices may be subject to local taxes which are calculated during checkout





Similar content being viewed by others
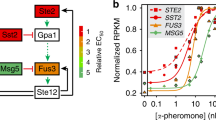
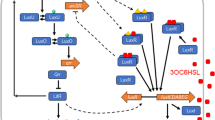
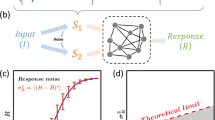
References
Dohlman, H. G. & Thorner, J. W. Regulation of G protein-initiated signal transduction in yeast: Paradigms and principles. Annu. Rev. Biochem. 70, 703–754 (2001)
Jackson, C. L. & Hartwell, L. H. Courtship in S. cerevisiae: Both cell types choose mating partners by responding to the strongest pheromone signal. Cell 63, 1039–1051 (1990)
Segall, J. E. Polarization of yeast cells in spatial gradients of alpha mating factor. Proc. Natl Acad. Sci. USA 90, 8332–8336 (1993)
Schrick, K., Garvik, B. & Hartwell, L. H. Mating in Saccharomyces cerevisiae: The role of the pheromone signal transduction pathway in the chemotropic response to pheromone. Genetics 147, 19–32 (1997)
Colman-Lerner, A. et al. Regulated cell-to-cell variation in a cell-fate decision system. Nature 437, 699–706 (2005)
Yi, T. M., Kitano, H. & Simon, M. I. A quantitative characterization of the yeast heterotrimeric G protein cycle. Proc. Natl Acad. Sci. USA 100, 10764–10769 (2003)
Cuatrecasas, P. Insulin–receptor interactions in adipose tissue cells: Direct measurement and properties. Proc. Natl Acad. Sci. USA 68, 1264–1268 (1971)
Kasai, M. & Changeux, J. P. In vitro excitation of purified membrane by cholinergic agonists. J. Membr. Biol. 6, 58–80 (1971)
Amir, S. M., Carraway, T. F., Kohn, L. D. & Winand, R. J. The binding of thyrotropin to isolated bovine thyroid plasma membranes. J. Biol. Chem. 248, 4092–4100 (1973)
Lin, S. Y. & Goodfriend, T. L. Angiotensin receptors. Am. J. Physiol. 218, 1319–1328 (1970)
Knauer, D. J., Wiley, H. S. & Cunningham, D. D. Relationship between epidermal growth factor receptor occupancy and mitogenic response. Quantitative analysis using a steady state model system. J. Biol. Chem. 259, 5623–5631 (1984)
Nagashima, T. et al. Quantitative transcriptional control of Erbb receptor signaling undergoes graded to biphasic response for cell differentiation. J. Biol. Chem. 282, 4045–4056 (2007)
Simons, S. S., Oshima, H. & Szapary, D. Higher levels of control: Modulation of steroid hormone-regulated gene transcription. Mol. Endocrinol. 6, 995–1002 (1992)
Rousseau, G. G. & Baxter, J. D. Glucocorticoid receptors. Monogr. Endocrinol. 12, 49–77 (1979)
Bloom, E. et al. Nuclear binding of glucocorticoid receptors: Relations between cytosol binding, activation and the biological response. J. Steroid Biochem. 12, 175–184 (1980)
Pedraza, J. M. & van Oudenaarden, A. Noise propagation in gene networks. Science 307, 1965–1969 (2005)
Black, H. S. Stabilized feed-back amplifiers. Electr. Eng. 53, 114–120 (1934)
Bhalla, U. S., Ram, P. T. & Iyengar, R. MAP kinase phosphatase as a locus of flexibility in a mitogen-activated protein kinase signaling network. Science 297, 1018–1023 (2002)
Black, J. W. & Leff, P. Operational models of pharmacological agonism. Proc. R. Soc. Lond. B 220, 141–162 (1983)
Savageau, M. A. Comparison of classical and autogenous systems of regulation in inducible operons. Nature 252, 546–549 (1974)
Becskei, A. & Serrano, L. Engineering stability in gene networks by autoregulation. Nature 405, 590–593 (2000)
Barkai, N. & Leibler, S. Robustness in simple biochemical networks. Nature 387, 913–917 (1997)
Gordon, A. et al. Single-cell quantification of molecules and rates using open-source microscope-based cytometry. Nature Methods 4, 175–181 (2007)
Gartner, A., Nasmyth, K. & Ammerer, G. Signal transduction in Saccharomyces cerevisiae requires tyrosine and threonine phosphorylation of Fus3 and Kss1. Genes Dev. 6, 1280–1292 (1992)
Tedford, K., Kim, S., Sa, D., Stevens, K. & Tyers, M. Regulation of the mating pheromone and invasive growth responses in yeast by two map kinase substrates. Curr. Biol. 7, 228–238 (1997)
Miyawaki, A. & Tsien, R. Y. Monitoring protein conformations and interactions by fluorescence resonance energy transfer between mutants of green fluorescent protein. Methods Enzymol. 327, 472–500 (2000)
van Drogen, F., Stucke, V. M., Jorritsma, G. & Peter, M. Map kinase dynamics in response to pheromones in budding yeast. Nature Cell Biol. 3, 1051–1059 (2001)
Bhattacharyya, R. P. et al. The ste5 scaffold allosterically modulates signaling output of the yeast mating pathway. Science 311, 822–826 (2006)
Maleri, S. et al. Persistent activation by constitutive Ste7 promotes Kss1-mediated invasive growth but fails to support Fus3-dependent mating in yeast. Mol. Cell. Biol. 24, 9221–9238 (2004)
Gruhler, A. et al. Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol. Cell. Proteomics 4, 310–327 (2005)
Shao, D., Zheng, W., Qiu, W., Ouyang, Q. & Tang, C. Dynamic studies of scaffold-dependent mating pathway in yeast. Biophys. J. 91, 3986–4001 (2006)
Bishop, A. C. et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature 407, 395–401 (2000)
Ballon, D. R. et al. DEP-domain-mediated regulation of GPCR signaling responses. Cell 126, 1079–1093 (2006)
Heximer, S. P. & Blumer, K. J. RGS proteins: Swiss army knives in seven-transmembrane domain receptor signaling networks. Sci. STKE 2007, pe2 (2007)
May, L. T., Leach, K., Sexton, P. M. & Christopoulos, A. Allosteric modulation of G protein-coupled receptors. Annu. Rev. Pharmacol. Toxicol. 47, 1–51 (2007)
Budas, G. R., Churchill, E. N. & Mochly-Rosen, D. Cardioprotective mechanisms of PKC isozyme-selective activators and inhibitors in the treatment of ischemia-reperfusion injury. Pharmacol. Res. 55, 523–536 (2007)
Lindsley, C. W., Barnett, S. F., Layton, M. E. & Bilodeau, M. T. The PI3K/Akt pathway: Recent progress in the development of ATP-competitive and allosteric Akt kinase inhibitors. Curr. Cancer Drug Targets 8, 7–18 (2008)
Shannon, C. A mathematical theory of communication. Bell Syst. Tech. J. 27, 379–423 (1948)
Bialek, W., Rieke, F., de Ruyter van Steveninck, R. R. & Warland, D. Reading a neural code. Science 252, 1854–1857 (1991)
Tkacik, G., Callan, C. & Bialek, W. Information flow and optimization in transcriptional regulation. Proc. Natl. Acad. Sci. U.S.A. 26, 12265–12270 (2008)
Gregor, T., Tank, D. W., Wieschaus, E. F. & Bialek, W. Probing the limits to positional information. Cell 130, 153–164 (2007)
Ausubel, F. M. et al. Current Protocols in Molecular Biology (John Wiley & Sons, Inc., 1987–2008)
Guthrie, C. & Fink, G. R. Methods in Enzymology, Guide to Yeast Genetics and Molecular Biology (Academic, 1991)
Brun, R., Couet, O., Vandroni, C. & Zanarini, O. PAW Physics Analysis Workstation CERN program library entry q121 (CERN, Geneva, 1989)
Jenness, D. D., Burkholder, A. C. & Hartwell, L. H. Binding of alpha-factor pheromone to Saccharomyces cerevisiae a cells: Dissociation constant and number of binding sites. Mol. Cell. Biol. 6, 318–320 (1986)
Bajaj, A. et al. A fluorescent α-factor analogue exhibits multiple steps on binding to its g protein coupled receptor in yeast. Biochemistry 43, 13564–13578 (2004)
Andersson, J., Simpson, D. M., Qi, M., Wang, Y. & Elion, E. A. Differential input by Ste5 scaffold and Msg5 phosphatase route a MAPK cascade to multiple outcomes. EMBO J. 23, 2564–2576 (2004)
Acknowledgements
We thank P. Abola, S. Andrews, A. Arkin, M. Bowen, L. Buck, C. Denby, A. Gann, D. Meldrum, T. Mitchison, C. Pabo, M. Ptashne, M. Reese, O. Resnekov, C. Ryan, M. Snyder, T. Thomson, A. E. Tsong and M. Wilson for discussions and/or comments on the manuscript, and O. Resnekov for help in articulating the requirements for fluidic induction devices. Work, including that of M.H. at the University of Washington, was supported by the Alpha Project at the Center for Quantitative Genome Function, an NIH Center of Excellence in Genomic Science under grant P50 HG02370 from the National Human Genome Research Institute to R.B.
Author Contributions R.C.Y. performed the Ste5 translocation, loss of G-protein and Dig1–Ste12 FRET, MAP kinase phosphorylation, mRNA measurements, and the image-based measurements of system output. C.G.P. and A.G. noted and helped articulate the relationship of negative feedback to dose–response overlap. C.G.P. and R.C.Y. performed flow cytometric measurements. L.L. and A.G. contributed to discussions about mutual information calculations and analysis. R.C.Y. and A.G. performed data analysis on loss of FRET experiments. A.C.-L. and A.G. carried out extensive initial measurements of Ste5 recruitment. K.B. provided unpublished information about protein quantification useful in initial discussions about dose–response alignment. R.C.Y. and D.P. performed numerous measurements verifying protein abundance in other strains. E.S. constructed the inhibitor-sensitive fus3-as2 and kss1-as2 alleles. M.H. designed and built fluidic devices used to induce the system in response to articulated requirements. R.B. provided input into project direction, experimental design and interpretation of results. R.C.Y., C.G.P. and R.B. wrote the paper and guarantee the integrity of the results.
Author information
Authors and Affiliations
Corresponding authors
Supplementary information
Supplementary Information
This file contains Supplementary Methods and Data, Supplementary Tables S1-S4 and S6, Supplementary Figures and Legends S1-S14 and Supplementary References (PDF 4551 kb)
Rights and permissions
About this article
Cite this article
Yu, R., Pesce, C., Colman-Lerner, A. et al. Negative feedback that improves information transmission in yeast signalling. Nature 456, 755–761 (2008). https://doi.org/10.1038/nature07513
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature07513
This article is cited by
-
Design principles of improving the dose-response alignment in coupled GTPase switches
npj Systems Biology and Applications (2023)
-
Stress response of Lymantria dispar asiatica (Lepidoptera: Erebidae) larvae and its gut microbiota to manganese ion
Journal of Forestry Research (2021)
-
Design of a MAPK signalling cascade balances energetic cost versus accuracy of information transmission
Nature Communications (2020)
-
Application of information theory in systems biology
Biophysical Reviews (2020)
-
Insights about collective decision-making at the genetic level
Biophysical Reviews (2020)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
