
Abstract
The genome of Aspergillus oryzae, a fungus important for the production of traditional fermented foods and beverages in Japan, has been sequenced. The ability to secrete large amounts of proteins and the development of a transformation system1 have facilitated the use of A. oryzae in modern biotechnology2,3,4. Although both A. oryzae and Aspergillus flavus belong to the section Flavi of the subgenus Circumdati of Aspergillus, A. oryzae, unlike A. flavus, does not produce aflatoxin, and its long history of use in the food industry has proved its safety. Here we show that the 37-megabase (Mb) genome of A. oryzae contains 12,074 genes and is expanded by 7–9 Mb in comparison with the genomes of Aspergillus nidulans5 and Aspergillus fumigatus6. Comparison of the three aspergilli species revealed the presence of syntenic blocks and A. oryzae-specific blocks (lacking synteny with A. nidulans and A. fumigatus) in a mosaic manner throughout the genome of A. oryzae. The blocks of A. oryzae-specific sequence are enriched for genes involved in metabolism, particularly those for the synthesis of secondary metabolites. Specific expansion of genes for secretory hydrolytic enzymes, amino acid metabolism and amino acid/sugar uptake transporters supports the idea that A. oryzae is an ideal microorganism for fermentation.
Similar content being viewed by others

Main
Sequencing of the A. oryzae genome was accomplished using the whole-genome shotgun (WGS) approach. The 37-Mb genome was predicted to contain a total of 12,074 genes encoding proteins with a length greater than 100 amino acid residues (see Methods). The genome was confirmed to comprise eight chromosomes (chromosomes 1–8 in decreasing size), the assignment of which is different from a previous report7 (Supplementary Table S1 and schematic drawing in Supplementary Fig. S1). Interestingly, the A. oryzae genome contained numerous stretches (1,750) of (A + T)-rich sequence (that is, >90% A + T composition in 50 nucleotides or longer), 6–9 times more than for A. fumigatus (197) and A. nidulans (308).
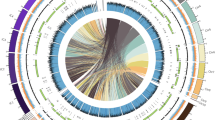
The A. oryzae genome is larger than those of A. fumigatus and A. nidulans by approximately 34% and 29%, respectively. Syntenic analysis of the three aspergilli revealed the presence of syntenic blocks and A. oryzae-specific blocks of sequence (lacking synteny with the two other aspergilli) in a mosaic manner throughout the A. oryzae genome (Fig. 1). Phylogenetic analysis of the three aspergilli using the whole-genome data showed that A. nidulans branched off earlier than A. oryzae and A. fumigatus5. Thus, the increase in genome size seems to be due to an A. oryzae lineage-specific acquisition of sequence, rather than loss of sequence in A. nidulans and A. fumigatus. If, on the other hand, A. nidulans and A. fumigatus are assumed to have lost 7–9 Mb of sequence after branching off from their A. oryzae-like ancestor, a greater proportion of syntenic blocks would be conserved between each of them and A. oryzae than between the two. However, we observed an almost equal proportion of syntenic blocks in the three species. This suggests that the genome size differences are largely due to sequence acquisition in A. oryzae. The expansion in genome size appears to be characteristic of the organisms closely related to A. oryzae, as the estimated genome size of its close relatives A. flavus (W. Nierman, personal communication) and Aspergillus niger8 is comparable to that of A. oryzae.

The blue bars at the bottom indicate the regions syntenic with A. fumigatus (AF) and A. nidulans (AN) genomes (see Methods). Non-metabolism, the genes relating to the COG categories other than metabolism; Q genes, secondary metabolism genes; Extra homologues, extra A. oryzae-specific homologues; AO-specific, A. oryzae-specific genes; Genes with EST(s), genes that have one or more corresponding EST(s); Gene density, distribution of all the predicted genes. Synteny was analysed as described in the Methods.
Using the cluster of orthologous group (COG)9 classification, most of the gene family expansion in the A. oryzae genome as compared to A. fumigatus was found to have occurred in those predicted to have roles in metabolism (C to Q), of which those for secondary metabolism (Q) are most significantly increased (Fig. 2; see also Supplementary Table S2). No significant differences were observed in the number of genes for any other COG category in comparison with A. nidulans and A. fumigatus, except for the genes involved in defence mechanisms (V) and extracellular structures (M).

The ratios of the number of genes in A. oryzae against those in A. fumigatus (AO/AF), A. nidulans (AO/AN), N. crassa (AO/NC) and S. cerevisiae (AO/SC) for each COG category9 were calculated. The ratio of the number of genes in A. nidulans against A. fumigatus (AN/AF) was also indicated. X indicates the genes without homology to any of the COG categories (see Methods). COGs with a gene number ≤5 for each species (Y, N and W) are not displayed to avoid misinterpretation derived from their possibly low reliability.
These secondary metabolism genes are enriched in regions lacking synteny with either A. fumigatus or A. nidulans (P = 9.8 × 10-32, see Fig. 1 for chromosome 1 and Supplementary Fig. S2 for all eight chromosomes), and the genes having expressed sequence tags (ESTs) are considerably enriched in the syntenic regions (P = 4.1 × 10-134). Many more cytochrome P450 genes were observed in A. oryzae (149) compared with A. nidulans (102) and A. fumigatus (65) (Table 1). Of the polyketide synthase (PKS) genes, a specific expansion of WA-like PKS genes was observed (Supplementary Table S3). In addition, the A. oryzae genome contained a variety of homologues of trichothecene hydroxylases, isotrichodermin hydroxylases and trichodiene oxygenases, as well as pisatin demethylases that are used by plant pathogenic fungi (for example, Nectria haematococca, Fusarium spp.) for detoxification of antimicrobial agents10. This is consistent with the close phylogenetic relationship of A. oryzae with the opportunistic plant pathogen A. flavus.
Although genes predicted to be involved in the aflatoxin synthetic pathway are present in A. oryzae, no ESTs of these genes were detected except for aflJ and norA (Akao, T. et al., unpublished data), whereas ESTs for all 25 of the aflatoxin pathway genes were found in A. flavus11. A. oryzae might have been selected as a non-toxigenic strain either during the long history of its industrial use or from the beginning.
In A. oryzae, all of the COG categories related to metabolism show an expansion of gene content (Fig. 2), the highest increase of which was observed for those involved in phenylalanine/tryptophan degradation (2 and 6 in Supplementary Fig. S3) and toluene/m-cresol/p-cymene degradation (9, 11 and 12 in Supplementary Fig. S3). This was based on the analysis using the Saccharomyces cerevisiae metabolic map as a reference. BAT1 and BAT2, which contribute to the metabolism of hydrophobic amino acids lysine and serine, are also over-represented (see Supplementary Fig. S4 for the entire metabolic pathways). There is also a significant expansion in the ATP-binding cassette (ABC), the amino acid-polyamine-organocation (APC) and the major facilitator superfamily (MFS) transporter genes (Supplementary Table S4), which are concerned with multidrug resistance, transport of amino acids and transport of sugars, respectively.
Within the koji culture, A. oryzae grows on the surface of solid material such as steamed rice or ground soybean, where amino acids and sugars are deficient at the beginning. The need for A. oryzae to get access to external nitrogen sources effectively and to degrade proteins and starches seems consistent with the observed expansion of the metabolism and transporter-related gene families. Judging from the EST data, the genes for alcohol dehydrogenase, pyruvate decarboxylase and sugar transporters are typical examples of the A. oryzae genes that are transcribed most strongly (Akao, T. et al., unpublished data). The strong expression of such genes might also have been enhanced through various adaptations12 during the course of domestication.
Aspergilli possess more sensor histidine kinases (13–15) than S. cerevisiae (1) and Schizosaccharomyces pombe (3), whereas histidine-containing phosphotransfer factors and response regulators are found in similar numbers. Aspergillus histidine kinases are classified into nine families (HK1–9), of which the HK8 orthologue is absent in Neurospora crassa and the sequenced plant pathogens Cochliobolus heterostrophus, Gibberella moniliformis, Fusarium graminearum and Magnaporthe grisea. Whereas A. fumigatus, A. nidulans, N. crassa and the plant pathogens possess a single HK6 gene (Nik-1 in N. crassa) that is essential for growth in high osmotic pressure, A. oryzae has two additional homologues. Continuous culturing under high osmolarity conditions (possibly through koji cultures) may have led to A. oryzae acquiring the additional Nik-1 homologues. There are three MAPKKs and MAPKKKs in the genomes of the three Aspergillus species and N. crassa. However, whereas A. nidulans and A. fumigatus possess four MAPKs and A. oryzae five, N. crassa, F. graminearum and M. grisea possess only three. Thus, A. oryzae may possess the most complex signal transduction cascade among the four filamentous fungi.
A. oryzae has the largest expansion of hydrolytic genes among the three aspergilli (Supplementary Table S5). The genomes of A. oryzae, A. fumigatus and A. nidulans contain 135, 99 and 90 secreted proteinase genes, respectively, which constitute roughly 1% of the total genes in each genome (Supplementary Table S6). All of the proteinase genes found in A. fumigatus and/or A. nidulans have orthologues in A. oryzae except for the one encoding aminopeptidase. On the other hand, several A. oryzae proteinase genes are missing in A. fumigatus and A. nidulans. Similarly, A. oryzae possesses more secretory proteinase genes that function in acidic pH, including aspartic proteinase, pepstatin-insensitive proteinase, serine type carboxypeptidase and aorsin (Supplementary Table S6). These increases may reflect A. oryzae's adaptation to acidic pH during the course of its domestication.
The phylogenetic tree of secretory aspartic proteinases from the three aspergilli genomes (Fig. 3) shows six homologous clusters (yellow boxes) distributed on all chromosomes other than chromosome 7. Their features, including intron conservation, are similar to each other except for cluster 4, which shows the highest diversity. Each cluster contains four member genes (blue boxes), namely three orthologues from each Aspergillus species and an extra A. oryzae-specific homologue. All of the extra A. oryzae homologues are located in the A. oryzae-specific regions, whereas the orthologous clusters are located in the common regions, except for AO070319000053 of cluster 4. It is interesting to note that the clustering feature of the orthologues and extra homologues for aspartic proteinases is also conserved with the genes for carboxypeptidases (Supplementary Fig. S5a) and metalloproteinases. In contrast, the number of genes encoding intracellular enzymes (Supplementary Fig. S5b), including serine proteinases, is consistent in the three aspergilli. A similar expansion pattern was also observed for the genes for maltases (Supplementary Fig. S5c) and extracellular α-glucosidases. Besides the secretory hydrolases, some metabolic genes, including those in glucose fermentation and lysine biosynthesis, showed a similar gene expansion pattern (Supplementary Fig. S6).

The phylogenetic relationship of aspartic proteinase homologues from the three aspergilli was analysed by the ClustalX30 program, successive unweighted pair-group method using arithmetic averages (UPGMA), and drawn by TreeView (Roderic, D. M., http://taxonomy.zoology.gla.ac.uk/rod/rod.html). Orange, blue and purple characters designate the A. oryzae, A. fumigatus and A. nidulans genes, respectively. Orthologous clusters among the three aspergilli and the clusters with an extra A. oryzae homologue are indicated by yellow and blue boxes, respectively.
It is well known that A. oryzae has three α-amylase genes (taka-amylase genes: amyA, amyB and amyC)13 that have almost identical nucleotide sequences with only one and two mismatches in the 5′-flanking and coding regions, respectively. The amyA gene has a transposon-like element at its 5′-flanking region, and the amyB and amyC genes have highly similar nucleotide sequences spanning approximately 5 kilobases (kb), including an incomplete transposon sequence at their 5′-flanking region. Phylogenetic analysis supports gene duplication to account for the expansion of the three α-amylase genes after A. oryzae branched off from the other two Aspergillus species (Supplementary Fig. S5d)—this is in clear contrast to the mode of gene expansion for the secretory proteinases mentioned above.
In contrast to the overall increase in the number of proteinases, A. oryzae has fewer glycosyl hydrolases with a cellulose-binding domain (five genes) or a starch-binding domain (glaA14) to digest insoluble cellulose or raw and granular starch, respectively (Supplementary Table S5). Apparently, no additional enzymes for accessing carbohydrates are required during fermentation in contrast to those, including knottins, found in A. fumigatus, which seems appropriate for its ecological niche of rotting vegetable matter.
Protein folding in the endoplasmic reticulum is assisted by chaperones (for example, BiP, calnexin) and foldases (three protein-disulphide isomerase family proteins and a peptidyl-prolyl cis–trans isomerase). As in other fungi, however, there is no calreticulin homologue (Supplementary Table S7). Major secretory component genes, which alter the efficiency of protein secretion, were identified in all three aspergilli with an exception of the A. fumigatus SSS1 homologue (Supplementary Table S8).
In comparison to the common regions, the A. oryzae-specific regions contained 1.7 times lower density of genes homologous to those in eukaryotes other than A. fumigatus and A. nidulans. In a search for bacterial homologues, we found two genes (AO070319000101 and AO070319000102) in an A. oryzae-specific region with highest sequence similarity to those of Agrobacterium tumefaciens (AGR_L_1864 (biotin carboxylase) and AGR_L_1866, hypothetical protein genes with E-values of 0.0 and 1 × 10-119, respectively). Because the two genes are adjacently located in both A. oryzae and A. tumefaciens (Supplementary Fig. S7a), and the two A. oryzae genes reside in a ‘bacterial cluster’ (Supplementary Fig. S7b), they are suggested to have been laterally transferred.
The expansion of A. oryzae-specific homologues might be the result of genome-wide duplication, as observed in yeast. The speciation of Aspergillus was estimated to have taken place approximately 20 million years ago15 and was later than the whole-genome duplication event in yeast, which was estimated to have taken place 150 million years ago16. We were unable to observe any extended stretch of region within the A. oryzae genome that showed a certain degree of similarity to another stretch of region despite the fact that we observed synteny among the three aspergilli (Fig. 1) and that segmentally duplicated stretches were detected by the same method within the S. cerevisiae genome. Thus, the increase in the genome size of A. oryzae relative to A. fumigatus and A. nidulans does not appear to be due to chromosomal duplication. The large segmental duplication, if any, must have taken place much earlier than the separation of the three aspergilli, and the similarity between the duplicated regions might have been completely lost by extensive sequence alterations and rearrangements. However, if the three aspergilli had a common ancestor possessing the expanded gene families found in A. oryzae, both A. nidulans and A. fumigatus must independently have lost approximately 3,000 genes in common with the putative common ancestor.
The mosaic structure of the genome, considered to be evidence for horizontal gene transfer17, was found by synteny analysis of the A. oryzae genome and was further characterized by the localization of the EST expression (see above) of non-metabolic genes (P = 1.78 × 10-95 and 1.32 × 10-51 for information and storage (J to B) and cellular function and signalling (D to O), respectively) and the genes of high codon adaptation index (top 5% genes, P = 9.8 × 10-28). The phylogenetic distance between the genes in the orthologous cluster and the A. oryzae-specific ones was similar to that between the genes of Aspergillus and the other genera belonging to Sordariomycetes. The statistical analysis by ref. 18 of some A. oryzae-specific homologues of aspartic proteinase, carboxypeptidase, maltase, pyruvate decarboxylase and lysine-ketoglutarate reductase/saccharopine dehydrogenase showed P-values of between 0.000 and 0.004. The results indicated phylogenetic inconsistency of these genes. These results, together with the above discussion, imply that the A. oryzae-specific genes have been transferred by a similar mechanism observed for an asexual pathogenic fungus19, in which chromosomes are transferred between genetically isolated clonal lines. It has been reported that yeast chromosomes are rearranged frequently under starved culture conditions and that (A + T)-rich sequences or transfer RNA often mediate such rearrangements20. Our EST analysis shows that the expression profile in solid-state cultivation is similar to that observed when a carbon source is omitted21. These reports suggest that the acquired foreign DNA has been rearranged in a short period of time by large-scale solid-state cultivation since A. oryzae was domesticated from an ancestor of A. flavus22.
It is tempting to speculate that the gene expansion of A. oryzae is explained by horizontal gene transfer; however, at this moment we cannot exclude the possibility of massive gene loss in the two other Aspergillus species. Future comparative analyses with more closely related species would provide more insight into the scenario of the genome evolution of A. oryzae, including that which occurred during the centuries of domestic cultivation.
Methods
Strain and DNA preparation
Aspergillus oryzae RIB40 (National Research Institute of Brewing Stock Culture and ATCC-42149) was used as the DNA donor. Genomic DNA preparation and removal of mitochondrial DNA was performed as described by refs 23 and 24, respectively.
Genome sequencing
The genome sequencing of A. oryzae was accomplished using the WGS approach by accumulating raw sequence reads of approximately ×9 depth of coverage. Contigs generated were mapped by Southern hybridization onto chromosomes separated by PFGE. Linkage between contigs was analysed by fingerprinting and PCR methods. Sequence assembly was validated with high-density end sequences of bacterial artificial chromosome (BAC) and cosmid clones and by Optical Mapping (OpGen). See Supplementary Information for details.
Gene prediction and annotation
Genes were predicted in the A. oryzae genome based on the homologies to known genes in the public database, ESTs of A. oryzae and A. flavus, and the statistical features of the genes by applying a combination of gene-finding software. Transfer RNAs were identified using tRNAScan-SE25. Repeated sequences were detected using RepeatMasker (Smit, A. F. A. and Green, P., http://ftp.genome.washington.edu/RM/RepeatMasker.html). The homologues of the proteins of aspergilli, N. crassa, M. grisea, Gibberella zeae, Penicillium and Paecilomyces are searched for by running BlastX with a threshold value of E ≤ 1 × 10-10. The resultant candidates of homologues are evaluated by ALN26, which predicts the precise gene structures by aligning the Blast hits and the protein sequences. ALN takes into account frameshift errors, coding potentials and signals for translational initiation, termination and splicing. Of the 6,586 genes thus predicted by ALN, 489 highly reliable genes were adopted into a learning set for GeneDecoder27 and GlimmerM28 software that work based on the statistic features of genes. GeneDecoder also integrates the information for splice sites provided by the ESTs, which are aligned with the genome sequence by SIM4 (ref. 29). Fivefold cross-validation of the gene finders trained by the above data set showed sensitivity/specificity for the exon prediction of 0.74/0.53 and 0.66/0.59 for GeneDecoder and GlimmerM, respectively, and those for coding sequences of 0.93/0.90 and 0.92/098. Genes partially supported by ESTs were predicted by GeneDecoder and those without any support by the known genes or ESTs were predicted by GlimmerM. The numbers of genes predicted by ALN, GlimmerM and GeneDecoder were 5,367, 6,983 and 1,713, respectively. All of the predicted protein-coding genes were annotated by searching against the COG database9 using BlastP, followed by manual corrections.
Synteny analysis
Orthologues between A. oryzae and either A. nidulans or A. fumigatus were identified using the best bi-directional hit method (BlastP with a bit score greater than 200). In addition, putative homologous regions between the species were identified by TBlastX with a bit score greater than 100. Orthologues and homologous regions between the contigs of two species were aligned to make a contiguous block, until no orthologues or homologous regions were found within the range of 10 kb. A region of conserved synteny was defined as the longest contiguous block that contained at least one orthologue and one additional orthologue or homologous region.
COG analysis
The number of genes for each COG category was analysed by a BlastP search using the amino acid sequences in the COG set9 with the bit score of ≥60.
Gene localization
Distribution of all predicted genes and the genes with ESTs that were obtained from mycelia grown in either liquid-rich medium, liquid-starved medium or solid-state cultivation (Akao, T. et al., unpublished data) were analysed by counting the corresponding genes in a 5-kb window. Distributions of non-metabolic genes, secondary metabolism genes, extra A. oryzae-specific homologues that have homology (bit score ≥100) to orthologues identified by best bi-directional match between A. oryzae and either A. fumigatus or A. nidulans, as well as A. oryzae-specific genes without homology to either A. fumigatus or A. nidulans genes (bit score <100) were analysed in the same way by applying a window size of 15 kb.
Statistical analyses
Localization of the secondary metabolism genes at the A. oryzae-specific regions was evaluated by the one-tailed P-value based on the binomial distribution with the sample size of 413. Localization of the genes with EST expression, non-metabolic genes and the top 5% of genes with a high CAI value at the syntenic regions was evaluated in the same way with sample sizes of 33,77, 1,839 and 703, respectively. The analyses were performed when A. oryzae-specific regions were detected by comparing the A. oryzae and A. fumigatus genomes. The phylogenetic inconsistency was statistically analysed by the method described in ref. 18 using data sets consisting of the genes of the three aspergilli and three species belonging to Sordariomycetes or Eurotiomycetes other than Aspergillus. The reference and test data sets included the A. oryzae gene in the orthologous cluster and the extra A. oryzae-specific homologue, respectively.
References
Gomi, K., Iimura, Y. & Hara, S. Integrative transformation of Aspergillus oryzae with a plasmid containing the Aspergillus nidulans argB gene. Agric. Biol. Chem. 51, 2549–2555 (1987)
Christensen, T. et al. High level expression of recombinant genes in Aspergillus oryzae. Bio/Technology 6, 1419–1422 (1988)
Ward, P. P. et al. Production of biologically active recombinant human lactoferrin in Aspergillus oryzae. Bio/Technology 10, 784–789 (1992)
Tsuchiya, K. et al. High level expression of the synthetic human lysozyme gene in Aspergillus oryzae. Appl. Microbiol. Biotechnol. 38, 109–114 (1992)
Galagan, J. E. et al. Sequencing of Aspergillus nidulans and comparative analysis with A. fumigatus and A. oryzae. Nature doi:10.1038/nature04341 (this issue)
Nierman, W. et al. Genomic sequence of the pathogenic and allergenic filamentous fungus Aspergillus fumigatus. Nature doi:10.1038/nature04332 (this issue)
Kitamoto, K., Kimura, K., Gomi, K. & Kumagai, C. Electrophoretic karyotype and gene assignment to chromosomes of Aspergillus oryzae. Biosci. Biotechnol. Biochem. 58, 1467–1470 (1994)
Archer, D. B. & Dyer, P. S. From genomics to post-genomics in Aspergillus. Curr. Opin. Microbiol. 7, 499–504 (2004)
Tatusov, R. L. et al. The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4, 41 (2003)
van den Brink, H. M., van Gorcom, R. F., van den Hondel, C. A. & Punt, P. J. Cytochrome P450 enzyme systems in fungi. Fungal Genet. Biol. 23, 1–17 (1998)
Yu, J., Whitelaw, C. A., Nierman, W. C., Bhatnagar, D. & Cleveland, T. E. Aspergillus flavus expressed sequence tags for identification of genes with putative roles in aflatoxin contamination of crops. FEMS Microbiol. Lett. 237, 333–340 (2004)
Ferea, T. L., Botstein, D., Brown, P. O. & Rosenzweig, R. F. Systematic changes in gene expression patterns following adaptive evolution in yeast. Proc. Natl Acad. Sci. USA 96, 9721–9726 (1999)
Sakaguchi, K., Takagi, M., Horiuchi, H. & Gomi, K. in Applied Molecular Genetics in Filamentous Fungi (eds Kinghorn, J. R. & Turner, G.) 54–99 (Blackie Academic & Professional, Glasgow, 1992)
Hata, Y. et al. Nucleotide sequence and expression of the glucoamylase-encoding gene (glaA) from Aspergillus oryzae. Gene 108, 145–150 (1991)
Hori, H. & Osawa, S. Origin and evolution of organisms as deduced from 5S ribosomal RNA sequences. Mol. Biol. Evol. 4, 445–472 (1987)
Langkjaer, R. B., Cliften, P. F., Johnston, M. & Piskur, J. Yeast genome duplication was followed by asynchronous differentiation of duplicated genes. Nature 421, 848–852 (2003)
Prade, R. A., Griffith, J., Kochut, K., Arnold, J. & Timberlake, W. E. In vitro reconstruction of the Aspergillus (= Emericella) nidulans genome. Proc. Natl Acad. Sci. USA 94, 14564–14569 (1997)
Lawrence, J. G. & Hartl, D. L. Inference of horizontal genetic transfer from molecular data: an approach using the bootstrap. Genetics 131, 753–760 (1992)
Masel, A. M., He, C., Poplawski, A. M., Irwin, J. A. G. & Manners, J. M. Molecular evidence for chromosome transfer between biotypes of Colletotrichum gloeosporioides. Mol. Plant–Microbe Interact. 9, 339–348 (1996)
Dunham, M. J. et al. Characteristic genome rearrangements in experimental evolution of Saccharomyces cerevisiae. Proc. Natl Acad. Sci. USA 99, 16144–16149 (2002)
Maeda, H. et al. Transcriptional analysis of genes for energy catabolism and hydrolytic enzymes in the filamentous fungus Aspergillus oryzae using cDNA microarrays and expressed sequence tags. Appl. Microbiol. Biotechnol. 65, 74–83 (2004)
Geiser, D. M., Pitt, J. I. & Taylor, J. W. Cryptic speciation and recombination in the aflatoxin-producing fungus Aspergillus flavus. Proc. Natl Acad. Sci. USA 95, 388–393 (1998)
Iimura, Y., Gomi, K., Uzu, H. & Hara, S. Transformation of Aspergillus oryzae through plasmid-mediated complementation of the methionine-auxotrohpic mutation. Agric. Biol. Chem. 51, 323–328 (1987)
Watson, J. & Thompson, W. F. Purification and restriction endonuclease analysis of plant nuclear DNA. Methods Enzymol. 118, 57–75 (1986)
Lowe, T. M. & Eddy, S. R. tRNAscan-SE: a program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 25, 955–964 (1997)
Gotoh, O. Homology-based gene structure prediction: simplified matching algorithm using a translated codon (tron) and improved accuracy by allowing for long gaps. Bioinformatics 16, 190–202 (2000)
Asai, K., Itou, K., Ueno, Y. & Yada, T. Recognition of human genes by stochastic parsing. Pac. Symp. Biocomput. 3, 228–239 (1998)
Majoros, W. H., Pertea, M., Antonescu, C. & Salzberg, S. L. GlimmerM, Exonomy and Unveil: three ab initio eukaryotic genefinders. Nucleic Acids Res. 31, 3601–3604 (2003)
Florea, L., Hartzell, G., Zhang, Z., Rubin, G. M. & Miller, W. A computer program for aligning a cDNA sequence with a genomic DNA sequence. Genome Res. 8, 967–974 (1998)
Thompson, J. D., Gibson, T. J., Plewniak, F., Jeanmougin, F. & Higgins, D. G. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res. 25, 4876–4882 (1997)
Acknowledgements
The authors are grateful to N. Hall and H. Hagiwara for discussions and critical reading of the manuscript. We thank M. Tadenuma and T. Ishikawa of the Brewing Society of Japan for the office work necessary for the collaborative research work of companies, national institutes and universities. Author Contributions M.S., T.T., K. Kusumoto, T.A., Y. Kashiwagi, H. Horikawa, A.H., R.I., Y. Kato, A.K., N.M., T.S., K.T., S.S., K. Isono, S.K., N.O., H.K. and M.M. sequenced the A. oryzae genome; the genes were computationally predicted and annotated from the A. oryzae genome by K.A., T. Kumagai, G.T., J.Y., D.B., T.E.C., O.G., T. Kin, H.N. and T. Komori; M.S., K. Kusumoto, T.A., O.A., Y. Kashiwagi, K.A., K.G., H. Horiuchi, K. Kitamoto, T. Kobayashi, M. Takeuchi, D.W.D., D.A., J.W.B., M.I., K. Iwashita, P.R.J., M.K., H.M., J.M., T.N., K. Oda, I.P., K.S., Y.T., O.Y., Y.Y., H.A., Y.H., Y. Koide, Y. Koyama, T.M., A.T. and M.M. contributed correction and hand annotation of the predicted genes; K.A., T. Kumagai, G.T., D.W.D., J.E.G., W.C.N., N.D.F., T. Kin, H.N., Y.T., J.W., T. Komori and M.M. analysed gene localization and development of the A. oryzae genome.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The genome sequence has been submitted to DDBJ under the accession numbers AP007150–AP007177. Reprints and permissions information is available at npg.nature.com/reprintsandpermissions. The authors declare no competing financial interests.
Supplementary information
Supplementary Methods
This describes the methods that were used for the library construction, sequencing, mapping contigs, southern hybridization and pulse field gel electrophoresis (DOC 44 kb)
Supplementary Figure 1
Schematic drawing of the A. oryzae genome sequence assembly. The 8 chromosomes of A. oryzae by the genome sequencing are represented from short arm to long arm together with the PFGE image of the A. oryzae chromosomes. (PPT 141 kb)
Supplementary Figure 2
Distribution and expression of genes throughout the A. oryzae genome. Non metabolism, Q genes, Extra homologues, AO specific genes, Genes with EST and Gene density are aligned with the eight A. oryzae chromosomes together with synteny to the A. fumigatus and A. nidulans genomes. (PDF 131 kb)
Supplementary Figure 3
Gene redundancy of pathway members. The amino acid sequences of S. cerevisiae genes for metabolic pathways members were obtained by SGD database and were searched by BLASTP with E-value of =< 10-20. (PDF 19 kb)
Supplementary Figure 4
Metabolic pathway view of the gene redundancy of pathway members. The gene redundancy of each member of the metabolic pathways in A. oryzae genome as compared to A. fumigatus and A. nidulans was analysed, and was superimposed on the metabolic pathway map. (PDF 100 kb)
Supplementary Figure 5
Phylogenetic analysis of proteinases and glucosidases. Phylogenetic relationship of carboxypeptidases, ATP-dependent proteinases, maltase and α-amylase genes from the three Aspergilli were analysed. (PDF 25 kb)
Supplementary Figure 6
Phylogenetic analysis of metabolic genes. Phylogenetic relationship of pyruvate decarboxylase, saccharopine dehydrogenase, homoaconitase and saccharopine dehydrogenase (NADP+, L-glutamate forming) in lysine biosynthesis from the three Aspergilli were analysed. (PDF 21 kb)
Supplementary Figure 7
Analysis of a pair of syntenic genes highly homologous between A. oryzae and A. tumefaciens. This figure shows the alignment of the pair of syntenic genes from the two species. Phylogenetic analysis using amino acid sequences of the genes from variety of organisms was carried out. (PDF 31 kb)
Supplementary Figures Legends
This file contains figure legends for Supplementary Figures 1–7. (DOC 37 kb)
Supplementary Tables
This file contains Supplementary Tables 1–8. (DOC 124 kb)
Rights and permissions
About this article
Cite this article
Machida, M., Asai, K., Sano, M. et al. Genome sequencing and analysis of Aspergillus oryzae. Nature 438, 1157–1161 (2005). https://doi.org/10.1038/nature04300
Received:
Accepted:
Issue Date:
DOI: https://doi.org/10.1038/nature04300
This article is cited by
-
Production of kojic acid by Aspergillus flavus OL314748 using box-Behnken statistical design and its antibacterial and anticancer applications using molecular docking technique
BMC Microbiology (2024)
-
Aspergillus oryzae PrtR alters transcription of individual peptidase genes in response to the growth environment
Applied Microbiology and Biotechnology (2024)
-
High-throughput droplet microfluidics screening and genome sequencing analysis for improved amylase-producing Aspergillus oryzae
Biotechnology for Biofuels and Bioproducts (2023)
-
Insights into the capability of the lignocellulolytic enzymes of Penicillium parvum 4-14 to saccharify corn bran after alkaline hydrogen peroxide pretreatment
Biotechnology for Biofuels and Bioproducts (2023)
-
Effective production of kojic acid in engineered Aspergillus niger
Microbial Cell Factories (2023)
Comments
By submitting a comment you agree to abide by our Terms and Community Guidelines. If you find something abusive or that does not comply with our terms or guidelines please flag it as inappropriate.
