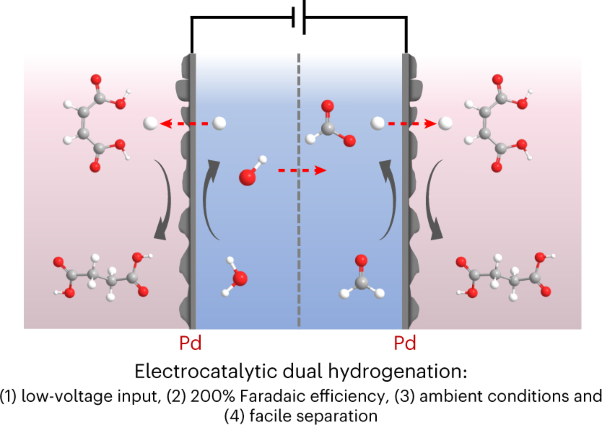
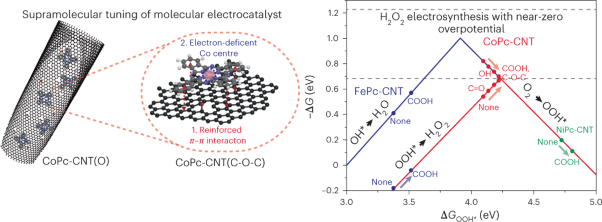
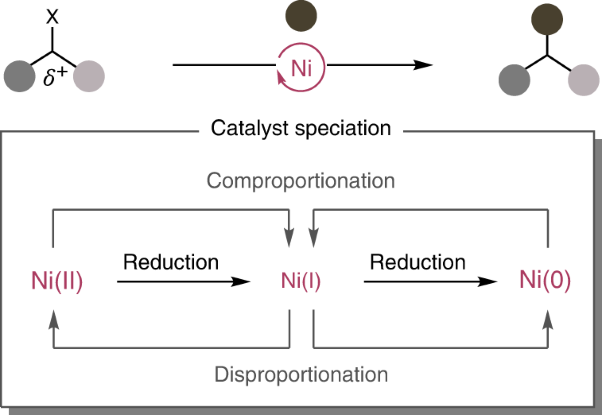
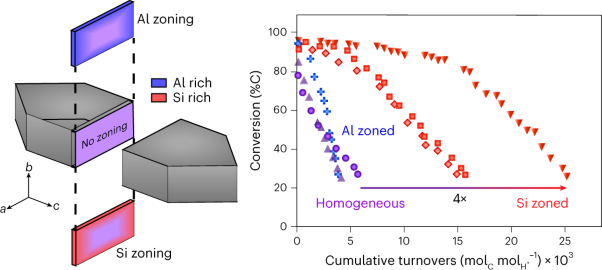
Volume 6 Issue 3, March 2023
Oxidoreductase at the tip
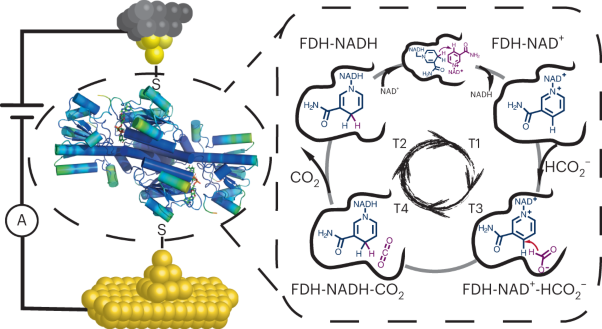
Formate dehydrogenase is immobilized on a scanning tunnelling microscope tip to measure the kinetic turnover of its enzymatic reaction showing that the bound NADH directly converts to NAD+ via in situ hydride exchange.
See Zhang et al.
Image: Wenjing Hong, Xiamen University. Cover design: Marina Spence.