
Abstract
Activating mutations in downstream genes of the epidermal growth factor receptor (EGFR) pathway may cause anti-EGFR resistance in patients with colorectal cancers. We present performance characteristics of a next-generation sequencing assay designed to detect such mutations. In this retrospective quality assessment study, we analyzed mutation detected in the KRAS, NRAS, BRAF, and PIK3CA genes by a clinically validated next-generation sequencing assay in 310 colorectal cancer specimens. Tumor cellularity and mutant allele frequency were analyzed to identify tumor heterogeneity and mutant allele-specific imbalance. Next-generation sequencing showed precise measurement of mutant allele frequencies and detected 23% of mutations with 2–20% mutant allele frequencies. Of the KRAS mutations detected, 17% were outside of codons 12 and 13. Among PIK3CA mutations, 48% were outside of codons 542, 545, and 1047. The percentage of tumors with predicted resistance to anti-EGFR therapy increased from 40% when testing for only mutations in KRAS exon 2 to 47% when testing for KRAS exons 2–4, 48% when testing for KRAS and NRAS exons 2–4, 58% when including BRAF codon 600 mutations, and 59% when adding PIK3CA exon 20 mutations. Right-sided colorectal cancers carried a higher risk of predicted anti-EGFR resistance. A concomitant KRAS mutation was detected in 51% of PIK3CA, 23% of NRAS, and 33% of kinase-impaired BRAF-mutated tumors. Lower than expected mutant allele frequency indicated tumor heterogeneity, while higher than expected mutant allele frequency indicated mutant allele-specific imbalance. Two paired neuroendocrine carcinomas and adjacent adenomas showed identical KRAS mutations, but only PIK3CA mutations in neuroendocrine carcinomas. Next-generation sequencing is a robust tool for mutation detection in clinical laboratories. It demonstrates high analytic sensitivity and broad reportable range, and it provides simultaneous detection of concomitant mutations and a quantitative measurement of mutant allele frequencies to predict tumor heterogeneity and mutant allele-specific imbalance.
Similar content being viewed by others


Main
Epidermal growth factor receptor (EGFR) regulates cell differentiation, proliferation, and survival via the mitogen-activated protein kinase (MAPK or RAS/RAF/MEK/ERK) pathway and the phosphatidylinositol 3-kinase (PI3K/AKT/mTOR) pathways.1 Cetuximab and panitumumab are anti-EGFR monoclonal antibodies approved by the Food and Drug Administration (FDA) for treatment of patients with KRAS-wild-type metastatic colorectal cancers, but the response rate is only ~10% in unselected patient populations.2, 3 This poor response to anti-EGFR therapy can be explained, in part, by activating mutations in genes of the downstream signaling pathways, such as KRAS or NRAS.4, 5, 6
In 2009, a provisional clinical opinion from the American Society of Clinical Oncology recommended testing for KRAS gene mutations in patients with metastatic colorectal cancers to predict response to anti-EGFR therapy.7 In addition to common KRAS mutations at codons 12 and 13, recent studies have shown that less common mutations at codons 59, 61, 117, and 146 and mutations of the NRAS gene also predict anti-EGFR resistance.5, 6, 7, 8, 9 BRAF mutations may also predict resistance to anti-EGFR therapy, although more prospective studies are needed to confirm these findings.5, 10 In colorectal cancers, mutations in the PIK3CA gene are usually accompanied by a KRAS mutation.5, 11 The benefit of testing PIK3CA mutations, particularly exon 20 mutations, to guide anti-EGFR therapy therefore requires further studies in a large cohort of colorectal cancer patients with wild-type KRAS and NRAS genes. The revised guideline for anti-EFGR therapy in 2014 from National Cancer Comprehensive Network recommends testing at least KRAS mutations at codons 12 and 13 as well as, whenever possible, additional KRAS mutations outside of exon 2, as well as NRAS mutations. It recommends considering BRAF testing if both KRAS and NRAS are nonmutated. The Association of Clinical Pathologists Molecular Pathology and Diagnostics Group in the United Kingdom also recommends that RAS analysis should include at least codons 12, 13, 59, 61, 117, and 146 of the KRAS gene and codons 12, 13, 59, and 61 of the NRAS gene.12
Massively parallel sequencing or next-generation sequencing technology has led to a revolution in genome discovery. The convergence of recent advances in molecular technology and rapid expansion of targeted therapeutics have changed approaches in clinical molecular diagnostics laboratories from the traditional ‘one test-one drug’ paradigm to the multiplexed genotyping platform that are used to simultaneously test a panel of genes for a specific tumor type.13, 14 Next-generation sequencing will soon become the most cost-effective multiplexed sequencing platform in the setting of clinical care as more and more biomarkers become mandatory clinical tests.13 Next-generation sequencing assays have also been retrospectively used to evaluate response to anti-EGFR therapy in colorectal cancer patients.8, 15 We have previously validated a next-generation sequencing platform using the AmpliSeq Cancer Hotspot Panel and Personal Genome Machine in a Clinical Laboratory Improvement Amendments-certified laboratory.16 In this retrospective analysis for quality assessment, we survey our experience with clinical mutation detection in KRAS, NRAS, BRAF, and PIK3CA genes in 310 colorectal cancer specimens using next-generation sequencing.
Materials and methods
A total of 314 formalin-fixed paraffin-embedded specimens with a diagnosis of colorectal cancer were submitted to the Molecular Diagnostics Laboratory at The Johns Hopkins Hospital between April 2013 and November 2014. The test was canceled in four specimens; two with scant tumor cells, one referral case with no tumor cells on the accompanying hematoxylin & eosin (H&E) slide, and one with the assay being successfully conducted in the primary tumor. The remaining 310 specimens were submitted from 305 tumors of 301 colorectal cancer patients (Supplementary Table S1). There were 258 resection specimens, 49 biopsy specimens, and three fine-needle aspiration specimens (Supplementary Table S2). Tissue blocks with adequate tumor cellularity were selected by the pathologists who made the diagnosis. Almost all were diagnosed as invasive adenocarcinoma, with the exceptions being one with neuroendocrine carcinoma (case 54) and one with mixed adenocarcinoma and neuroendocrine carcinoma (case 282). One H&E slide followed by 5–10 unstained slides and one additional H&E slide were prepared with PCR precautions. The H&E slide was examined and marked by pathologists for subsequent macro-dissection of formalin-fixed paraffin-embedded neoplastic tissues from 3–10 unstained slides of 5 or 10-μm-thick sections. DNA was isolated from the area(s) designated by pathologists using the Pinpoint DNA Isolation System (Zymo Research, Irvine, CA, USA), followed by further purification via the QIAamp Mini Kit (Qiagen, Valencia, CA, USA).17 The estimated percentage of tumor nuclei was separated into 5 quintiles (1–20%, 21–40%, 41–60%, 61–80%, and 81–100%) by two pathologists (GZ and MTL). In the presence of discrepancy, the mean value was applied.
Next-generation sequencing was conducted using AmpliSeq Cancer Hotspot Panel (v2) for targeted multi-gene amplification, as described previously.16 Briefly, we used the Ion AmpliSeq Library Kit 2.0 for library preparation, Ion OneTouch 200 Template Kit v2 DL and Ion OneTouch Instrument for emulsion PCR and template preparation, and the Ion Personal Genome Machine 200 Sequencing Kit with the Ion 318 Chip and Personal Genome Machine as the sequencing platform (Life Technologies, Carlsbad, CA, USA). The DNA input was up to 30 ng, as measured by Qubit 20 Fluorometer (Life Technologies). Up to eight specimens were barcoded using Ion Xpress Barcode Adapters (Life Technologies) for each Ion 318 chip. One to three controls (a non-template control, a normal peripheral blood control from a male, and/or positive control specimens) were included in each chip. Positive controls were mixed DNA specimens from several cell lines with known mutations (Supplementary Table S3).
Mutations were identified and annotated through both Torrent Variant Caller (Life Technologies) and direct visual inspection of the binary sequence alignment/map file using the Broad Institute's Integrative Genomics Viewer. All specimens were analyzed for BRAF, KRAS, NRAS, and PIK3CA genes. During our validation of this next-generation sequencing assay, a cutoff of background noise at 2% was chosen for single-nucleotide variations according to a study of 16 non-neoplastic formalin-fixed paraffin-embedded tissues.16 We also developed a statistical model to determine the read depth needed for a given percent tumor cellularity and number of functional genomes. With sufficient DNA input, the limit of detection is dictated by the depth of coverage (or number of sequencing reads). Approximately 150 and 500 reads is needed to detect a heterozygous mutation at a 99% confidence in a specimen with 20 and 10% tumor cellularity, respectively. The reportable ranges and reference ranges are summarized in Supplementary Table S4.
Pyrosequencing and SNP array analysis was performed as previously described.18 Correlation between % KRAS mutant versus % PIK3CA mutant was examined by Spearman’s rank correlation coefficient (denoted as r) using the GraphPad Prism software (GraphPad Software, ver5, La Jolla, CA, USA).
Results
Positive and Negative Controls
No mutation was detected in 88 runs of the negative control specimen and all mutations in the positive control specimens were detected. The observed mutant allele frequencies were highly consistent throughout the test period, demonstrating that next-generation sequencing is a precise quantification assay for mutant allele frequency (Figure 1 and Supplementary Table S3).

Precision of mutant allele quantification by next-generation sequencing. Mutant allele percentage detected in a positive control specimen was highly consistent across 38 runs.
KRAS, NRAS, BRAF, and PIK3CA Mutations in Colorectal Cancers
Two of 310 specimens failed (0.6%), including one of two specimens from different blocks of a tumor. Five paired specimens from the same tumor showed identical mutation status. Next-generation sequencing data, therefore, were available for analysis in 304 tumors (Supplementary Table S1). Mutations were not detected in any of the KRAS, NRAS, BRAF, and PIK3CA genes (quadruple mutation-negative) in 98 (32%) tumors. One mutation was detected in 167 tumors, two mutations in 36 tumors, and three mutations in three tumors. Because our analysis pipeline included both Torrent Variant Caller and direct visual inspection of all reported amplicons using Integrative Genomics Viewer, three mutations (8.8% PIK3CA p.V344M [c.1030G>A], 1.95% KRAS p.Q61H [c.183A>C], and 40% BRAF p.Y472C [c.1415A>G]) were detected by Integrative Genomics Viewer inspection but missed by Torrent Variant Caller.
Next-generation sequencing was retrospectively performed in two specimens that had been canceled because of insufficient tumor cells on the H&E slides. DNA was extracted from 7 to 10 unstained slides and next-generation sequencing assays conducted with 0.8 and 1.8 ng DNA, respectively. One specimen showed 19% KRAS p.A146T (c.436G>A), 34% PIK3CA p.V344M (c.1030G>A), and 43% PIK3CA p.H1047R (c.3140A>G) mutations and the other a 37% KRAS p.G12C (c.34G>T) mutation. The overall assay success rate was 310 out of 312 (99.4%).
A total of 20 unique KRAS mutations were detected in 146 out of 304 tumors (48%), including one tumor with both p.G12A (c.35G>C) and p.G13D (c.38G>A) mutations. The mutations were located in exon 2 in 123 tumors (41%), exon 3 in 10 tumors (3.3%) and exon 4 in 13 tumors (4.3%). p.G12D (c.35>G>A), p.G12V (c.35G>T), and p.G13D (c.38G>A) were the most common KRAS mutations in colorectal cancers (Supplementary Figure S1). Mutations located outside codon 12 or 13 were observed in 25 tumors (17% of KRAS positive tumors). Six unique NRAS mutations were detected in 13 tumors (4.3%), 5 at codon 12 or 13 and 8 at codon 61. Five unique BRAF mutations were detected in 36 tumors (12%), including 28 p.V600E (c.1799T>A) mutations with predicted highly elevated BRAF kinase activity, 2 p.N581S mutations with unknown kinase activity, and six mutations (4 p.D594G [c.1781A>G], one p.D594N [c.1780G>A] and one p.Y472C [c.1415A>G]) with predicted reduced or silent kinase activity.19, 20
Twenty-three unique PIK3CA mutations were detected in 47 tumors (16%), including five tumors with double PIK3CA mutations and 17 tumors with mutations detected only in the PIK3CA gene. The PIK3CA exon 20 mutation, most associated with resistance to EGFR therapy, without concomitant KRAS, NRAS, and BRAF mutations was observed in only five tumors (1.6%). Most mutations were located within exons 9 (42%), 20 (23%), and 1 (17%) (Supplementary Figure S2). All but p.E110del (c.328_330del) and p.E453del (c.1359_1361del) mutations were single-base substitution, including a p.E81A (c.242A>C) mutation not reported in the COSMIC database (last access on 14 May 2015). The three most common codons (E542, E545, and H1047) account for only 27 of 52 (52%) PIK3CA mutations.
Prediction of Resistance to Anti-EGFR Therapy in the Right- and Left-Sided Colorectal Cancers
Cancers proximal or distal to the splenic flexure were classified as right-sided or left-sided. The primary tumors were located within the right colon in 116 (38%) tumors and the left colorectum in 176 (58%) tumors. The exact location of the primary tumor was not known in nine tumors and the exact location of the primary tumors within the transverse colon was not known in three tumors. Quadruple mutation-negative tumors were observed in only 16 right-sided colorectal cancers and in 78 left-sided colorectal cancers (14 vs 44%, P<0.001) (Table 1). Right-sided colorectal cancers showed a significantly higher incidence of mutations in BRAF, KRAS, NRAS genes within the MAPK pathway (Table 1). Cecal cancer exhibited the highest frequency of RAS mutations (63 vs 51% of non-cecum right-sided colorectal cancers, 41% of non-rectum left-sided colorectal cancers, and 47% of rectal cancers) and the lowest frequency of quadruple mutation-negative tumors (12 vs 19% of non-cecum right-sided colorectal cancers, 48% of non-rectum left-sided colorectal cancers, and 42% of rectal cancers).
Resistance to anti-EGFR therapy has been associated with mutations at codons 12, 13, 59, 61, 117, and 146 of the KRAS and/or NRAS gene, and to a lesser extent, p.V600E mutations of the BRAF gene and exon 20 mutations of the PIK3CA gene.4, 5, 6 The predicted population with lack of response increased from 40% when testing for only mutations in KRAS exon 2 to 47% when testing for KRAS exons 2–4, 48% when testing for KRAS and NRAS exons 2–4, 58% when including BRAF codon 600 mutations, and 59% when adding PIK3CA exon 20 mutations (Figure 2). The right-sided colorectal cancers carry a higher risk of containing resistance mutations to anti-EGFR therapy (79 vs 49%, P<0.001). The difference between right-sided and left-sided colorectal cancers was not significant when only KRAS codons 12 and 13 mutations were examined (43 vs 36%, P=0.25).

Prediction of resistance to anti-epidermal growth factor receptor therapy (y axis) according to reportable ranges of assays (x axis). KRAS*: codons 12, 13, 59, 61, 117, and 146 of the KRAS gene; RAS*: codons 12, 13, 59, 61, 117, and 146 of the KRAS and NRAS genes; BRAF*: codon 600 of the BRAF gene.
Mutant Allele Frequency as Quality Assessment Tool to Identify Tumor Heterogeneity
A total of 251 mutations were detected in the 308 specimens with next-generation sequencing data. The mutant allele frequency was 2–5% in six out of 251 mutations (2.4%), 5–10% in 14 mutations (5.6%), and 10–20% in 38 mutations (15%). Among the 20 specimens with 10% or fewer mutant alleles, the mutant allele percentage was consistent with a 1–20% or 11–30% estimated tumor cellularity in 12 specimens (Table 2). These included one lymph node specimen with an infiltrative metastasis (case 1) (Supplementary Figure S3a), three small biopsy specimens (cases 67, 174, and 227) and one resected specimen (case 76) with tumor cells surrounded by prominent desmoplastic reaction and/or inflammatory cell infiltration (Supplementary Figure S3b), one resected mucinous adenocarcinoma (case 270) (Supplementary Figure S3c), and five resected specimens of rectal cancer (cases 75, 98, 158, 217, and 287) and one resected specimen of liver metastasis (case 213) with prior chemo/radiotherapy and/or embolization therapy (Supplementary Figure S3d).
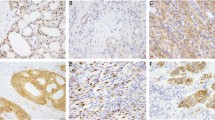
Mutations with an observed allele frequency at least 10 percentage points lower than that expected from the estimated tumor cellularity were observed in eight specimens, suggesting that these mutations are present in a subpopulation of the neoplasm. DNA samples isolated from several subpopulations within tumors demonstrated intra-tumor heterogeneity for the KRAS mutation in case 33 (Supplementary Figure S4) and the presence of NRAS and PIK3CA mutations in different mesentery tumor nodules in case 127 (Supplementary Figure S5). In case 282 with a cecal mixed adenocarcinoma and neuroendocrine carcinoma, a 53% KRAS p.A146T (c.436G>A) mutation and an 8.9% PIK3CA p.E545K (c.1633G>A) were detected in the originally isolated DNA specimen extracted from a designated area containing mainly the adjacent adenoma and a small component of the invasive neuroendocrine carcinoma (Figures 3 and 4). The PIK3CA mutation was detected only in the DNA re-isolated from the invasive neuroendocrine carcinoma, but not the adenoma (Figure 4).

Invasive neuroendocrine carcinoma (b) and adjacent adenoma (a) in case 282. Original magnifications of the hematoxylin and eosin stained slides are × 10 for (a) and × 40 for (b).

PIK3CA mutation in invasive neuroendocrine carcinoma but not adenoma. In case 282, tubular adenoma showed only a KRAS p.A146T (c.436G>A) mutation (a and c), while the invasive neuroendocrine carcinoma (NE CA) showed a KRAS p.A146T mutation and a PIK3CA p.E545K (c.1633G>A) mutation (b and d). Mutant allele frequency is shown in the parenthesis.
Concomitant Mutations
Concomitant mutations within KRAS, NRAS, BRAF, and PIK3CA genes were observed in 39 out of 206 tumors (19%) with mutations (Table 3). These included one tumor with two different KRAS mutations, three tumors with a KRAS mutation and two PIK3CA mutations, two tumors with two PIK3CA mutations, and 33 tumors with two mutations of different genes. Tumors with a PIK3CA mutation showed a significantly higher incidence of concomitant mutations of different genes (64%) as compared with tumors with a KRAS (21%) or BRAF (19%) mutation (P<0.001) (Supplementary Figure S6). Concomitant KRAS mutation was seen in 24 out of 47 (51%) PIK3CA-mutated tumors, three out of 13 (23%) NRAS-mutated tumors, and two out of six BRAF codon 594 or 472 mutated tumors. Concomitant mutations with KRAS, BRAF, or NRAS mutations were detected in 18 out of 22 (82%) tumors with a PIK3CA exon 9 mutation and in seven out of 12 (58%) tumors with a PIK3CA exon 20 mutation.
PIK3CA Mutation in the Invasive Neuroendocrine Carcinoma but Not the Adjacent Adenoma
In addition to case 282, DNA samples were also isolated from adjacent adenomas in four specimens with concomitant KRAS and PIK3CA mutations. The same mutations were detected in the adenoma and the originally examined invasive adenocarcinoma of cases 76, 106, and 108 (Table 4). Similarly to case 182, only a KRAS mutation was observed in the adenoma of case 54, even though both KRAS and PIK3CA mutations were detected in the originally examined invasive neuroendocrine carcinoma.
Prediction of Mutant Allele-Specific Imbalance Using Concomitant Mutant Allele Percentages
The extremely high concordance of mutant allele frequencies (r=0.99) in five tumors with two PIK3CA mutations and one tumor with two KRAS mutations suggests that the paired mutations of the same gene most likely occurred within the same tumor cells (Supplementary Figure S7). The KRAS and PIK3CA mutant allele frequencies in 24 tumors with concomitant mutations were also significantly correlated, but the degree of association was weaker (r=0.56) (Figure 5). The ratio of PIK3CA/KRAS mutant allele frequencies was within the range between 80 and 120% in only 10 tumors and was below 80% (17–67%) in 12 tumors. A PIK3CA mutant allele frequency lower than the KRAS mutant allele frequency suggests that (i) the PIK3CA mutation was present in a subpopulation of the tumor cells (as shown in case 282) or (ii) a higher incidence of amplification of the KRAS mutant allele or loss of the wild-type allele of the KRAS gene. This was confirmed by SNP array analysis from five invasive tumors showing no aneuplody of chromosome 3q containing the PIK3CA genes and gain of chromosome 12p12 containing the KRAS gene (Table 4 and Supplementary Figure S8).

Correlation of mutant allele frequencies in tumors with concomitant KRAS and PIK3CA mutations. The lines labeled with 80 and 120% indicate the boundary of events with the PIK3CA/KRAS mutant allele ratio between 80 and 120%. Only one PIK3CA mutant allele was included for the three specimens with a KRAS mutation and two PIK3CA mutations. r: Spearman’s rank correlation coefficient.
Discussion
A variety of molecular assays have been developed for clinical detection of KRAS and BRAF mutations. Specimens containing low tumor cellularity are not uncommon in the clinical diagnostic setting.21, 22 With an analytic sensitivity of 10–20% mutant allele frequency, the prior gold standard of Sanger sequencing would have missed 20 out of 251 (8%) mutations with <10% mutant allele frequency or 58 (23%) mutations with <20% mutant allele frequency in this series. This is particularly problematic for colorectal cancer specimens with prior neoadjuvant therapy.22 The analytic sensitivity can be improved to ~5% mutant allele frequency by using pyrosequencing, high-resolution melting analysis or real-time PCR assays or even 1% with allele-specific PCR assays.18, 23, 24, 25 Cobas 4800 is a real-time PCR assay approved by the FDA of the United States for companion BRAF testing. The Therascreen KRAS RGQ PCR kit is an allele-specific PCR assay approved as a companion KRAS test.23, 24, 25 However, cobos 4800 detects only the p.V600E mutation and non-p.V600E codon 600 mutations at a lower analytic sensitivity. Therascreen detects seven common KRAS mutations in 8 PCR reactions and would have missed 26 of 147 (18%) of KRAS mutations detected by next-generation sequencing in this study. In addition, these assays detect only one or a few hotspots in a single run and may not be suitable for some core biopsy or fine-needle aspiration specimens containing limited tissue.
In an era of continuous expansion of predictive markers for targeted therapeutics, multiplexed assays are becoming popular in clinical laboratories. Primer extension-based assays with a multiplex design, such as the Sequenom MassARRAY system, detect multiple hotspots in a single reaction while retaining an analytic sensitivity of 5% mutant alleles or less.5, 26 In this study, we demonstrated that next-generation sequencing provides not only a high analytic sensitivity (low limit of detection) but also a broad reportable range for simultaneous mutation detection of a panel of KRAS, NRAS, and potentially BRAF and PIK3CA genes relevant to anti-EGFR therapy of colorectal cancers in a clinical diagnostic setting. Twenty-five out of 147 (17%) KRAS mutations were located outside codons 12 and 13 and eight out of 36 (22%) BRAF mutations were located outside codon 600. Only 52% of PIK3CA mutations occurred at codons 542, 545, and 1047 with our next-generation sequencing assay. Application of assays with broader reportable ranges may shed light on the clinical significance of uncommon mutations. For example, response to anti-EGFR therapy has been observed in a patient with a p.D594G mutation, which encodes a BRAF protein with impaired kinase activity.5, 19 BRAF mutations with reduced or silent kinase activity do not directly activate the downstream MAPK pathway.19, 20 In the presence of oncogenic RAS proteins, however, kinase-silent BRAF forms a complex with CRAF and leads to hyperactivation of the CRAF/MEK/ERK cascade.20 BRAF codon 594 mutations have been associated with a higher incidence of concomitant RAS mutations (4 in 34, 12%).20 In our colorectal cancer population, concomitant KRAS mutations were observed in two out of six cases with BRAF mutations (one at codon 472 and five at codon 594) with impaired kinase activity.19, 27
One study has suggested that PIK3CA exon 20 mutations may be predictive for anti-EGFR resistance,5 but their significance is still debated, partly because of a low incidence of PIK3CA-only mutations.11 In two recent meta-analyses of KRAS wild-type patients, only 12–14 patients with a PIK3CA exon 20 mutation could be enrolled for analysis.28, 29 In this study, only five (1.6%) tumors were positive for PIK3CA exon 20 mutation without concomitant KRAS, NRAS, and BRAF mutations. Like PIK3CA exon 20-only mutations, NRAS-only mutations were observed in only 4 (1.3%) tumors.
Common mutations of KRAS, NRAS, BRAF, and PIK3CA genes were examined using the Sequenom MassARRAY system in 773 patients treated with cetuximab.5 The conditional inference tree analysis for response suggested that KRAS testing should be considered first, BRAF second, NRAS third, and PIK3CA exon 20 fourth. The response rate improved from 24% in an unselected population to 41% in the quadruple negative population, which accounted for 50% of the population. Using next-generation sequencing assays, which have a broader reportable range, only 32% of tumors (14% of right-side colorectal cancers and 44% of left-side colorectal cancers) were quadruple mutation-negative. The predicted anti-EGFR-resistant population increased from 40% when testing only KRAS codons 12 and 13 to 59% when KRAS and NRAS codons 12, 13, 59, 61, 117, and 146; BRAF codon 600; and PIK3CA exon 20 were included. However, more prospective studies are needed to elucidate the benefit of testing BRAF and PIK3CA mutations to guide anti-EGFR therapy.
Proximal and distal colorectal cancers differ in terms of clinicopathological features and molecular oncogeneisis.30, 31, 32 BRAF mutations are predominantly observed in right-sided colorectal cancers regardless of microsatellite instability status.31, 33 Association of KRAS mutations with tumor location remains controversial. Most large studies of more than 1000 colorectal cancers showed consistently higher incidences of KRAS codons 12 and 13 mutations in the right-sided colorectal cancers (38–40 vs 30–35% similar to 43 vs 36% in this study), albeit with a much less substantial difference than that seen in BRAF mutations.33, 34, 35, 36 In the current study with broader reportable ranges of the next-generation sequencing assay, right-sided colorectal cancers showed a higher incidence of mutations in KRAS, NRAS, and BRAF genes within the MAPK pathway. The observation that RAS mutations occur at a higher frequency in cecal cancers compared with colorectal cancers in other locations has also been reported previously.34, 36 These results were consistent with the gene expression profiles showing activated signature of the MAPK pathway in the right-sided colorectal cancers and the clinical study showing a greater progress-free survival benefit from cetuximab therapy in left-sided colorectal cancer patients with wild-type KRAS status.31, 37
Consistent mutant allele frequencies over a 20-months period in positive control specimens highlighted again the precise quantitative nature of the next-generation sequencing assays.21 During our validation of this clinical assay, we also showed that mutant allele frequencies of KRAS and BRAF mutations detected by pyrosequencing and next-generation sequencing were highly concordant, suggesting that next-generation sequencing assays are also accurate in their quantification.18, 38 We have also shown that correlation of the observed mutant allele frequency and the expected mutant allele frequency based on the estimated tumor cellularity may serve as a measure for post-analytic quality assessment.18, 22 The discrepancy between observed and expected mutant allele frequencies may result from tumor heterogeneity, mutant allele-specific imbalance, assay bias and/or inaccurate estimation of tumor cellularity.
Mutations occurring after initiation of the founder clone may be present in only a subclonal population. Whether these mutations could be detected depends on the analytic sensitivity of assays used and the portion of tumor examined. Mutations may be present in a subclonal population when the observed mutant allele frequency is lower than that expected from the tumor cellularity (Table 2). This was confirmed in the current study by examining separate areas of neoplasm. Detection of KRAS or NRAS mutations in a subclonal population of colorectal cancers may be clinically informative. In colorectal cancers, tumors with ‘secondary’ KRAS or NRAS mutations may arise from a small subpopulation present within the original tumor before treatment or as a consequence of continued mutagenesis over the course of anti-EGFR treatment.39, 40 Presence of KRAS-mutant subclones has also shown correlation with worse progression free survival in colorectal cancer patients treated with anti-EGFR therapy.32
Because it is quantitative, next-generation sequencing also provides the relative mutant allele frequencies of concomitant mutations. Discrepancy of mutant allele frequency in concomitant mutations is most likely caused by mutant allele-specific imbalance, but it may also indicate that one mutant is present in a subclonal population. In this study, the same KRAS mutations were observed in two pairs of invasive neuroendocrine carcinomas and adjacent adenomas, but only invasive tumors showed a PIK3CA mutation. This explains only an 8.9% PIK3CA mutation in the original sampling of both adenoma and invasive neuroendocrine carcinoma with an overall 61–80% estimated tumor cellularity in case 282. These results also indicate that the invasive neuroendocrine carcinoma is derived from the adenoma after gain of the PIK3CA mutation, similar to the stepwise genetic alteration model associated with colorectal tumorigenesis. This model proposes that PIK3CA mutations occur after KRAS and BRAF mutations and, in cooperation with other oncogenes or tumor suppressor genes, drive the clonal evolution from large adenoma to invasive adenocarcinoma.41
Next-generation sequencing demonstrates a high analytic sensitivity, broad reportable range of mutation spectrum, capacity for quantitative measurement of mutant allele frequencies, and simultaneous detection of concomitant mutations. Mutant allele frequency and tumor cellularity can be used as a quality assessment measure to predict and identify tumor heterogeneity and mutant allele-specific imbalance. Further studies are warranted to elucidate the clinical and/or biological significance of uncommon mutations, concomitant mutations, mutant allele-specific imbalance, and anti-EGFR resistant mutations detected in minor subclonal populations.
References
Yarden Y, Sliwkowski MX . Untangling the ErbB signalling network. Nat Rev Mol Cell Biol 2001;2:127–137.
Cunningham D, Humblet Y, Siena S et al. Cetuximab monotherapy and cetuximab plus irinotecan in irinotecan-refractory metastatic colorectal cancer. N Engl J Med 2004;351:337–345.
Van Cutsem E, Peeters M, Siena S et al. Open-label phase III trial of panitumumab plus best supportive care compared with best supportive care alone in patients with chemotherapy-refractory metastatic colorectal cancer. J Clin Oncol 2007;25:1658–1664.
Karapetis CS, Khambata-Ford S, Jonker DJ et al. K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 2008;359:1757–1765.
De Roock W, Claes B, Bernasconi D et al. Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy-refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 2010;11:753–762.
Douillard JY, Oliner KS, Siena S et al. Panitumumab-FOLFOX4 treatment and RAS mutations in colorectal cancer. N Engl J Med 2013;369:1023–1034.
Allegra CJ, Jessup JM, Somerfield MR et al. American Society of Clinical Oncology provisional clinical opinion: testing for KRAS gene mutations in patients with metastatic colorectal carcinoma to predict response to anti-epidermal growth factor receptor monoclonal antibody therapy. J Clin Oncol 2009;27:2091–2096.
Peeters M, Oliner KS, Parker A et al. Massively parallel tumor multigene sequencing to evaluate response to panitumumab in a randomized phase III study of metastatic colorectal cancer. Clin Cancer Res 2013;19:1902–1912.
Sorich MJ, Wiese MD, Rowland A et al. Extended RAS mutations and anti-EGFR monoclonal antibody survival benefit in metastatic colorectal cancer: a meta-analysis of randomized, controlled trials. Ann Oncol 2015;26:13–12.
Di Nicolantonio F, Martini M, Molinari F et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 2008;26:5705–5712.
Cathomas G . PIK3CA in colorectal cancer. Front Oncol 2014;4:35.
Wong NA, Gonzalez D, Salto-Tellez M et al. RAS testing of colorectal carcinoma—a guidance document from the Association of Clinical Pathologists Molecular Pathology and Diagnostics Group. J Clin Pathol 2014;67:751–757.
Dienstmann R, Rodon J, Barretina J et al. Genomic medicine frontier in human solid tumors: prospects and challenges. J Clin Oncol 2013;31:1874–1884.
Deeb KK, Sram JP, Gao H et al. Multigene assays in metastatic colorectal cancer. J Natl Compr Canc Netw 2013;11:S9–17.
Ciardiello F, Normanno N, Maiello E et al. Clinical activity of FOLFIRI plus cetuximab according to extended gene mutation status by next-generation sequencing: findings from the CAPRI-GOIM trial. Ann Oncol 2014;25:1756–1761.
Lin MT, Mosier SL, Thiess M et al. Clinical validation of KRAS, BRAF, and EGFR mutation detection using next-generation sequencing. Am J Clin Pathol 2014;141:856–866.
Lin MT, Tseng LH, Rich RG et al. Δ-PCR, a simple method to detect translocations and insertion/deletion mutations. J Mol Diagn 2011;13:85–92.
Dudley JC, Gurda GT, Tseng LH et al. Tumor cellularity as a quality assurance measure for accurate clinical detection of BRAF mutations in melanoma. Mol Diagn Ther 2014;18:409–418.
Wan PT, Garnett MJ, Roe SM et al. Mechanism of activation of the RAF-ERK signaling pathway by oncogenic mutations of B-RAF. Cell 2004;116:855–867.
Heidorn SJ, Milagre C, Whittaker S et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 2010;140:209–221.
Frampton GM, Fichtenholtz A, Otto GA et al. Development and validation of a clinical cancer genomic profiling test based on massively parallel DNA sequencing. Nat Biotechnol 2013;31:1023–1031.
Dudley J, Tseng LH, Rooper L et al. Challenges posed to pathologists in the detection of KRAS mutations in colorectal cancers. Arch Pathol Lab Med 2015;139:211–218.
Anderson S, Bloom KJ, Vallera DU et al. Multisite analytic performance studies of a real-time polymerase chain reaction assay for the detection of BRAF V600E mutations in formalin-fixed, paraffin-embedded tissue specimens of malignant melanoma. Arch Pathol Lab Med 2012;136:1385–1391.
Halait H, Demartin K, Shah S et al. Analytical performance of a real-time PCR-based assay for V600 mutations in the BRAF gene, used as the companion diagnostic test for the novel BRAF inhibitor vemurafenib in metastatic melanoma. Diagn Mol Pathol 2012;21:1–8.
Harbison CT, Horak CE, Ledeine JM et al. Validation of companion diagnostic for detection of mutations in codons 12 and 13 of the KRAS gene in patients with metastatic colorectal cancer: analysis of the NCIC CTG CO.17 trial. Arch Pathol Lab Med 2013;137:820–827.
Paik PK, Arcila ME, Fara M et al. Clinical characteristics of patients with lung adenocarcinomas harboring BRAF mutations. J Clin Oncol 2011;29:2046–2051.
Sen B, Peng S, Tang X et al. Kinase-impaired BRAF mutations in lung cancer confer sensitivity to dasatinib. Sci Transl Med 2012;4:136ra70.
Huang L, Liu Z, Deng D et al. Anti-epidermal growth factor receptor monoclonal antibody-based therapy for metastatic colorectal cancer: a meta-analysis of the effect of PIK3CA mutations in KRAS wild-type patients. Arch Med Sci 2014;10:1–9.
Mao C, Yang ZY, Hu XF et al. PIK3CA exon 20 mutations as a potential biomarker for resistance to anti-EGFR monoclonal antibodies in KRAS wild-type metastatic colorectal cancer: a systematic review and meta-analysis. Ann Oncol 2012;23:1518–1525.
Iacopetta B . Are there two sides to colorectal cancer? Int J Cancer 2002;101:403–408.
Missiaglia E, Jacobs B, D'Ario G et al. Distal and proximal colon cancers differ in terms of molecular, pathological, and clinical features. Ann Oncol 2014;25:1995–2001.
Loupakis F, Yang D, Yau L et al. Primary tumor location as a prognostic factor in metastatic colorectal cancer. J Natl Cancer Inst 2015;107:dju427.
Roth AD, Tejpar S, Delorenzi M et al. Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60-00 trial. J Clin Oncol 2010;28:466–474.
Yamauchi M, Morikawa T, Kuchiba A et al. Assessment of colorectal cancer molecular features along bowel subsites challenges the conception of distinct dichotomy of proximal versus distal colorectum. Gut 2012;61:847–854.
Gonsalves WI, Mahoney MR, Sargent DJ et al. Patient and tumor characteristics and BRAF and KRAS mutations in colon cancer, NCCTG/Alliance N0147. J Natl Cancer Inst 2014;106:dju106.
Rosty C, Young JP, Walsh MD et al. Colorectal carcinomas with KRAS mutation are associated with distinctive morphological and molecular features. Mod Pathol 2013;26:825–834.
Brule SY, Jonker DJ, Karapetis CS et al. Location of colon cancer (right-sided [RC] versus left-sided [LC]) as a predictor of benefit from cetuximab (CET): NCIC CTG CO.17. J Clin Oncol 2013;31:3528.
Chen G, Dudley J, Tseng LH et al. Lymph node metastases of melanoma: challenges for BRAF mutation detection. Hum Pathol 2015;46:113–119.
Diaz LA Jr, Williams RT, Wu J et al. The molecular evolution of acquired resistance to targeted EGFR blockade in colorectal cancers. Nature 2012;486:537–540.
Misale S, Yaeger R, Hobor S et al. Emergence of KRAS mutations and acquired resistance to anti-EGFR therapy in colorectal cancer. Nature 2012;486:532–536.
Jones S, Chen WD, Parmigiani G et al. Comparative lesion sequencing provides insights into tumor evolution. Proc Natl Acad Sci US A 2008;105:4283–4288.
Acknowledgements
Supported by 1UM1CA186691-01 from the National Cancer Institute of USA.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Additional information
Supplementary Information accompanies the paper on Modern Pathology website
Rights and permissions
About this article

Cite this article
Haley, L., Tseng, LH., Zheng, G. et al. Performance characteristics of next-generation sequencing in clinical mutation detection of colorectal cancers. Mod Pathol 28, 1390–1399 (2015). https://doi.org/10.1038/modpathol.2015.86
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2015.86
This article is cited by
-
The gene mutational discrepancies between primary and paired metastatic colorectal carcinoma detected by next-generation sequencing
Journal of Cancer Research and Clinical Oncology (2018)
-
Current applications of molecular pathology in colorectal carcinoma
Applied Cancer Research (2017)
-
Clinical detection and categorization of uncommon and concomitant mutations involving BRAF
BMC Cancer (2015)
