
Abstract
Systemic chemotherapies for advanced or metastatic thyroid carcinomas have been of only limited effectiveness. For patients with differentiated or medullary carcinomas unresponsive to conventional treatments, novel therapies are needed to improve disease outcomes. Multiple novel therapies primarily targeting angiogenesis have entered clinical trials for metastatic thyroid carcinoma. Partial response rates up to 30% have been reported in single-agent studies, but prolonged disease stabilization is more commonly observed. The most successful agents target the vascular endothelial growth factor receptors, with potential targets including the mutant kinases associated with papillary and medullary oncogenesis. Two drugs approved for other malignancies, sorafenib and sunitinib, have had promising preliminary results reported, and are being used selectively for patients who do not qualify for clinical trials. At least one randomized, placebo-controlled phase III trial has been successfully completed, showing improved progression-free survival in patients with advanced or metastatic medullary thyroid carcinoma treated with vandetanib. Randomized trials for other agents are currently underway. Treatment for patients with metastatic or advanced thyroid carcinoma now emphasizes clinical trial opportunities for novel agents with considerable promise. Alternative options now exist for use of tyrosine kinase inhibitors that are well tolerated and may prove worthy of regulatory approval for this disease.
Similar content being viewed by others

Main
Thyroid carcinoma is a malignancy of rapidly increasing importance in public health as well as research into novel therapeutics. Across the world, the incidence of the disease has been steadily rising.1 In the United States, the age-adjusted incidence of thyroid carcinoma increased by an annual average 6.1% between 1998 and 2007, and age-adjusted mortality increased by an annual average of 0.9% during the same time period.2 Given that relative survival from thyroid cancer markedly declines after age 65, and the doubling of the population greater than 65 years old expected in the next 20 years, mortality from thyroid cancer can only be expected to rise further unless significant improvements in treatment of metastatic disease can be achieved.3, 4
The treatment of most patients with differentiated thyroid carcinoma (DTC; both papillary (PTC) and follicular (FTC) histologies) is based on surgery, radioactive iodine, and thyroid hormone therapy.5 When metastatic disease occurs, radioactive iodine can be curative. In fact, the first reported instance of cure of a patient with metastatic cancer was in a man with metastatic FTC treated with four doses of radioactive iodine.6 It was soon recognized, however, that radioiodine would only be curative in a minority of patients with metastatic disease, often due to ‘the spotty nature of iodine concentration’.7, 8, 9 TSH-suppressive thyroid hormone therapy can help to slow the pace of the disease, as TSH can activate its receptor on the cancer cell and promote tumor growth and proliferation.10 However, for those patients with metastatic DTC that progresses despite radioiodine and TSH-suppressive thyroid hormone therapy, no effective therapy exists.
Treatment of patients with medullary thyroid carcinoma (MTC) is also based on surgery for primary and regional metastatic disease. Because the neuroendocrine-derived MTC is not responsive to either radioiodine or TSH suppression, these options are not available for treatment of progressive, metastatic MTC. The outcomes of patients with metastatic disease are no better than those with radioiodine-unresponsive DTC.11
Cytotoxic systemic chemotherapies for advanced, metastatic thyroid carcinomas have limited effectiveness, with response rates typically 25% or less.12 With such poor outcomes, results from few clinical trials of new therapies for thyroid carcinomas were published during the latter half of the twentieth century. Plaguing these early trials was the practice of lumping patients with all histologies of thyroid carcinoma, confounding interpretation of the results. The definitions of response used in these earlier studies varied as well, and none is comparable to the currently used standard methodology.13, 14 Thus, treatment with cytotoxic chemotherapy is generally limited to patients with symptomatic or rapidly progressive metastatic disease unresponsive to or unsuitable for surgery, radioiodine (for tumors derived from differentiated carcinomas), and external beam radiotherapy.
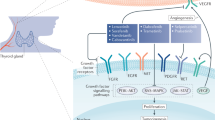
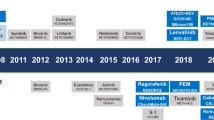
During the past decade, biologic discoveries have sparked trials testing novel, biologically targeted therapies for advanced thyroid carcinomas. Of prime importance has been recognition of key oncogenic mutations in PTC and MTC. BRAF and RAS genes code for kinases that activate signaling through the mitogen-activated protein kinase (MAPK) pathway, regulating growth and function in many cells both normal and neoplastic. Evidence from various tumor models supports the contention that most PTCs arise as a result of single activating somatic mutations in one of three genes: BRAF, RAS, and translocations producing RET/PTC oncogenes.15 The resultant RET/PTC proteins signal upstream from RAS, thus activating the same MAPK pathway. The presence of BRAF mutations is associated with decreased expression of mRNAs for proteins that yield differentiated functions of follicular cells, such as the sodium iodide symporter and the TSH receptor.16 Clinically, BRAF mutations are associated with radioiodine unresponsiveness and increased rates of disease recurrence and mortality.17, 18 For MTC, almost all familial forms of the disease arise because of inheritable germ-line activating mutations in RET, and identical somatic mutations occurring in C cells commonly cause sporadic disease. Activated RET mutant proteins also enhance MAPK signaling. Consistent with the ‘oncogene addiction’ hypothesis, inhibition of these etiologic activating mutations leads to either tumor stabilization or regression. Therefore, interest arose in the therapeutic potential of kinase inhibitors for these diseases.
A second development was recognition of processes facilitating tumor growth, reflecting either normal (such as hypoxia-inducible angiogenesis) or abnormal (such as epigenetic modifications of chromosomal DNA and histones) adaptations. Angiogenesis is critical in supporting tumor cell growth and metastasis, supplying nutrients and oxygen, removing waste products, and facilitating distant metastasis.19 Of the identified proangiogenic factors, vascular endothelial growth factor (VEGF) is key, binding to two receptor tyrosine kinases, VEGFR-1 (Flt-1) and VEGFR-2 (Flk-1/KDR), that also trigger MAPK signaling.20 In PTC, the intensity of VEGF expression correlates with a higher risk of metastasis and recurrence, a shorter disease-free survival, and BRAF mutation status.21, 22, 23
This review will focus on findings from key studies that reflect this new paradigm for research-driven treatment using targeted therapies for metastatic thyroid carcinoma.24 (Online databases that can be searched to identify clinical trials currently recruiting patients can be found at www.thyroid.org and www.clinicaltrials.gov).
Targeting oncogenic kinases
Given the oncogenic roles of activated BRAF, RET, and RET/PTC kinases, it is theorized that specific targeting of these kinases could block tumor growth and induce senescence.25 To date, only selective inhibitors of BRAF have entered clinical trials as a test of this hypothesis, as the agents available to target RET and RET/PTC generally also inhibit VEGFR and other kinases.26 In contrast with the experience of treating gastrointestinal stromal tumors containing activating c-KIT mutations with the KIT-inhibitor imatinib, use of selective BRAF inhibitors has not yet yielded impressive results in BRAF-mutant PTC.27 The emerging evidence of a high frequency of squamous cell neoplasms as an adverse event seen with all BRAF inhibitors may reveal a novel mechanism of oncogenesis.
PLX 4032
PLX 4032 is an orally available small molecule that specifically inhibits only the V600E mutant BRAF kinase, without significant impairment of wild-type BRAF or other RAF kinases.28 In melanoma and colon carcinoma cell lines bearing the V600E BRAF mutation, the IC50 values for inhibiting phosphorylation of ERK were 10–30 nM and for inhibiting cellular proliferation were 47–126 nM.28, 29 The RET/PTC mutant thyroid cancer cell line TPC1, however, was poorly inhibited, with an IC50 for cellular proliferation of 10 μM. In contrast, BRAF mutant cell lines are effectively inhibited at concentrations <100 nM, inducing a cell-cycle blockade but not leading to cell death.30 Preliminary data from a phase I study of escalating doses of PLX 4032 described the outcomes of three patients with BRAF-mutant PTC.29 One PTC patient experienced a partial response with shrinkage of lung metastases, whereas the other two patients had prolonged stable disease. Among the overall cohort of 55 patients with solid tumors (49 of whom had melanoma), the most common adverse events were skin rash, fatigue, pruritus, photosensitivity, and nausea. Although severe side effects were uncommon, 11% of the patients developed cutaneous squamous cell carcinomas.
XL281
XL281, an oral small molecule that inhibits both wild-type and mutant BRAF kinases at low nanomolar concentrations, is currently in phase I trial.31 Preliminary data described prolonged stable disease in six PTC patients; of the two patients whose tumors were documented to contain BRAF mutations, both remained stable after more than 1 year of therapy, as did a third PTC patient whose mutation status was unknown. Another patient with Hurthle cell carcinoma was also treated with prolonged stable disease. No partial response was observed in any of the thyroid cancer patients. The most common side effect reported among all 48 solid tumor patients in the trial was fatigue in nearly half of patients, and other common toxicities included nausea, diarrhea, and vomiting, all of which were occasionally severe. Four patients were also described as having developed either cutaneous squamous cell carcinomas or premalignant keratoacanthomas.
Targeting signaling kinases
A wide variety of multitargeted kinase inhibitors have entered clinical trials for patients with advanced or progressing metastatic thyroid cancers. Most of these agents have had the common property of inhibiting VEGF receptors at nanomolar concentrations, and thus have targeted angiogenesis primarily. However, given the considerable structural similarity between RET and VEGFR kinases, most of these small-molecule inhibitors are capable of affecting both kinases. Because of the targeting similarities of many of these agents, common toxicities exist among these agents, including hypertension, diarrhea, skin rashes, and fatigue.
Motesanib
Motesanib (AMG706) is an oral, tyrosine kinase inhibitor targeting the VEGF receptors 1, 2, and 3.32 In both in vitro and cell-based assays, nanomolar concentrations of motesanib inhibited autophosphorylation of both wild-type and mutant RET; growth of xenografts of TT cells bearing the C634W RET mutation was effectively inhibited.33 In a phase I study, motesanib showed antitumor activity in patients with advanced solid malignancies, including five patients with DTC and one with MTC; three thyroid patients experienced >30% reductions in tumor diameters, qualifying as partial responders.13, 34 The most common toxicities included fatigue, nausea, diarrhea, and hypertension, all typical of this class of drugs.
On the basis of this phase I experience, a multicenter, open-label phase II trial was initiated, testing the efficacy of motesanib in separate cohorts of patients with progressive DTC35 and patients with progressive or symptomatic MTC,36 starting at 125 mg daily. The eligibility criterion of progression was based upon serial radiographic imaging studies within the preceding 6 months, applying RECIST response assessment.13 Of 93 DTC patients who initiated therapy, one-third were still on drug after 48 weeks. Partial response was confirmed by subsequent imaging and independent radiologic review in 14% of the DTC patients, and another 35% of these previously progressive disease patients maintained stable disease for at least 24 weeks. The median progression-free survival (PFS) was 40 weeks. Although the drug does not inhibit BRAF, patients with BRAF mutation-bearing tumors were less likely to progress while on drug, which may relate to higher dependence upon VEGF-mediated angiogenesis in such tumors.37 Of 91 patients with progressive or symptomatic MTC who initiated therapy, only 2% had confirmed partial response but another 47% experienced stable disease for at least 24 weeks.36 Unexpectedly, the maximum and trough plasma concentrations of the drug in MTC patients were lower than reported with other solid tumor patients, and these differing pharmacokinetics may have contributed to the lower response rate. In an analysis of biomarkers, a greater than 5.3-fold increase in concentration of placental growth factor after 3 weeks of therapy was predictive of an increased likelihood of objective response across the two cohorts.38 Overall, the drug was well tolerated, with similar side effects as reported in the phase I trial. An unanticipated side effect of motesanib therapy was a 30% increase in the mean dosages of levothyroxine required to maintain TSH suppression or euthyroidism, respectively, in DTC and MTC cohorts, and 60–70% of patients experienced peak TSH concentrations out of the therapeutic ranges.39
Sorafenib
Sorafenib (BAY 43-9006) is an oral, small-molecule TKI targeting VEGF receptors 2 and 3, RET (including most mutant forms that have been examined), and BRAF.40 In preclinical studies, sorafenib prevented the growth of the TPC1 and TT cell lines, which contain the RET/PTC1 and C634W RET mutations, respectively.41 In four phase I trials testing varying doses and administration schedules of sorafenib, the optimal therapeutic dose was found to be 400 mg twice daily.42 The most common or significant toxicities included hand–foot syndrome, rash, fatigue, diarrhea, and hypertension. Like other agents that inhibit BRAF, sorafenib also has been associated with development of cutaneous squamous cell carcinomas in up to 5% of treated patients, and a similar frequency of keratoacanthomas and other premalignant actinic lesions.43
Although no thyroid cancer patients were reported in these phase I trials, tumor shrinkage was reported in one thyroid cancer patient included in a phase II trial for advanced solid tumors.44 Subsequently, three phase II trials have been performed focusing on patients with metastatic DTC, collectively representing the largest cohort of thyroid cancer patients studied with any single chemotherapy agent.
-
A phase II trial recruited 58 patients in a 10-month period.45 Although RAI treatment failure was required, demonstration of tumor progression was not an entry requirement. Of 41 PTC patients, confirmed partial response was observed in 15% (with a median time to response of at least 1 year), and stable disease was described in another 61%. For the subgroup of PTC patients whose cancer had not previously been treated with chemotherapy, median PFS was 16 months. Another 11 patients had FTC or Hurthle cell carcinoma; no objective responses were observed, and median PFS was only 4.5 months.
-
In a smaller phase II study, unconfirmed partial responses were reported in 4 of 15 (27%) evaluable patients with PTC and 3 of 7 with FTC (43%).46 Median PFS was 84 weeks. Updated follow-up data from this latter trial were recently presented, comprising 55 carcinomas (25 with PTC, 19 with FTC or HTC, 4 with MTC, and 5 with poorly differentiated or anaplastic carcinomas).47 Although the overall PFS remained 84 weeks, it was significantly shorter at 54 weeks in those patients whose tumors lacked the BRAF activating mutation.
-
A phase II study aimed to evaluate the effect of 26 weeks of sorafenib therapy on radioiodine uptake and tumor response in 32 patients with progressive, radioiodine-negative DTC.48 At study end, 8 (25%) patients had a partial response, 11 had stable disease (34%), 7 had progressive disease (22%), and 6 were nonevaluable. Median PFS was 58 weeks, although patients with bone metastases had worse median PFS than those without (47 weeks compared with 69 weeks, P<0.05). Of 21 patients who underwent radioiodine imaging after 26 weeks of treatment, none had induction of uptake in metastatic lesions. The most commonly reported and serious adverse events included hand–foot syndrome, weight loss, hypertension, diarrhea, alopecia, rash, mucositis, and hypocalcemia. One patient experienced a myocardial infarction, and another congestive heart failure.
In a recent retrospective series, sorafenib therapy was associated with prolongation of median PFS by at least 1 year, compared with patients' rate of disease progression before initiation of therapy.49 A randomized, placebo-controlled phase III study of sorafenib as first-line therapy for progressive metastatic DTC has been initiated.
Anticipating synergy between sorafenib's ability to inhibit MAPK signaling and the RAS-blocking effects of the farnesyltransferase inhibitor tipifarnib, a phase I trial was performed of the combination of these drugs. The maximum tolerated doses of sorafenib and tipifarnib were 200 and 100 mg twice daily, respectively. In the 22 patients with DTC treated, median PFS was 20 months.50
The anti-RET activity of sorafenib makes MTC a potential therapeutic target for this drug as well.51 In a small pilot study, five patients with metastatic MTC were treated with sorafenib, starting at 400 mg twice daily.52 Responses were described in two (including one complete response) after 6 months of treatment and symptomatic improvement was observed in all, but most patients required dose reduction due to side effects. Preliminary results have been reported from larger, open-label phase II study in patients with metastatic MTC.53 Although partial response was observed in only 6% of patients with sporadic MTC, stable disease lasting more than 6 months was reported in 62%. A high frequency of side effects was noted, including flushing, diarrhea, weight loss, alopecia, hand–foot syndrome, and rash. Severe adverse events included a pulmonary embolus, hypokalemia, hypertension, hyponatremia, joint pain, and thrombocytopenia. Partial responses were also reported in four of nine evaluable MTC patients participating in the phase I study of the combination of sorafenib and tipifarnib.50
In anaplastic carcinoma cell lines, preclinical models suggested potential efficacy of sorafenib to inhibit MAPK signaling.54 Subsequently, a phase II trial was started, evaluating sorafenib therapy in patients who had progressed after previous cytotoxic chemotherapy.55 Of 15 patients evaluated, 2 had experienced a partial response and 4 had stable disease as their best responses to treatment, but the overall median time to progression was only 1.5 months and duration of survival was 3.5 months.
Sorafenib is approved by the US Food and Drug Administration as treatment for advanced renal cell carcinoma and unresectable hepatocellular carcinoma. Although not specifically approved for thyroid carcinomas, sorafenib is being used in selected patients with progressive metastatic papillary and MTC for whom clinical trials are not appropriate.56 The drug may also be appropriate in selected pediatric cases; in one report, treatment with sorafenib yielded a marked response in a child whose lung metastases from PTC were progressing despite radioiodine therapy.57 As with other antiangiogenic therapies, pediatric usage may result in bony growth plate inhibition and growth abnormalities.
Sunitinib
Sunitinib (SU11248) is an oral, small-molecule TKI of all three VEGF receptors, RET, and RET/PTC subtypes 1 and 3.58 Prolonged partial responses have been described in three patients (with PTC, FTC, and MTC, respectively) treated with sunitinib, 50 mg daily for 28 days followed by 14 days of no treatment per cycle.59, 60 FDG uptake by positron emission tomography imaging was markedly reduced in the DTC patients. Preliminary results from an open-label phase II trial in patients with progressive DTC or MTC report partial response in 13% of 31 DTC patients, and disease stabilization in 68% of DTC and 83% of MTC patients.61 Common or severe adverse events include fatigue, diarrhea, palmar-plantar erythrodysesthesia, neutropenia, and hypertension. Interim analysis from a second open-label phase II trial reported partial responses or stable disease for more than 12 weeks in 2 of 12 DTC and 3 of 8 MTC patients.62 Recently, preliminary results from a third trial, using a lower dose of 37.5 mg daily but administered continuously, were reported.63 Of 33 patients with FDG-PET avid metastatic thyroid cancer (26 with DTC, 7 with MTC), 29 were evaluable for response: 7% complete response (lasting at least 9 months), 25% partial response, and 48% stable disease. Like sorafenib, sunitinib is approved for treatment of renal cell carcinoma, and is therefore available for use in selected thyroid cancer patients with metastatic disease warranting therapy outside clinical trials.
Vandetanib
Vandetanib (ZD 6474) is an oral, small-molecule TKI that targets VEGF receptors 2 and 3, RET, and at higher concentrations, the EGF receptor.64, 65 One of the first small-molecule inhibitors to be studied in thyroid cancer cell lines, vandetanib was shown to inhibit effectively RET/PTC3 mutations found in some PTC and M918T RET mutations occurring in MEN2B-associated and some sporadic MTC.66 Growth of cell lines containing RET/PTC1 or RET/PTC3 was inhibited. However, the drug was not able to block RET when a hydrophobic amino-acid substitution occurs at V804, as in some inherited forms of MTC.67 In a phase I trial in 77 patients with various solid carcinomas other than thyroid, doses up to 300 mg daily were tolerated with the most common dose-limiting side effects of diarrhea, hypertension, and skin rash.68
On the basis of the preclinical demonstration that vandetanib inhibited most RET point mutations, a multicenter, open-label phase II trial studied the efficacy of the drug in patients with metastatic familial forms of MTC.69 Thirty patients were enrolled, starting therapy with vandetanib, 300 mg daily. Confirmed partial response was reported in 21% of these patients, and unconfirmed responses in another 17%. Calcitonin levels dropped by more than 50% in most patients, but blocking RET may lead to a direct inhibition of calcitonin gene expression, independent of tumor volume changes.70 The most commonly reported side effects included rash (particularly photosensitivity), diarrhea, fatigue, and nausea, whereas the most severe toxicities included asymptomatic QT interval prolongation, rash, and diarrhea. A second phase II trial in familial MTC, starting at 100 mg daily, reported similar results.71
A multicenter, randomized phase III trial of vandetanib compared with placebo in patients with locally advanced or metastatic MTC was preliminarily reported.72 With a median follow-up of 24 months, median PFS was markedly improved (hazard ratio 0.45 (95% confidence interval 0.30–0.69). No difference was observed in overall survival between the two treatment arms despite the improvement in PFS. Although patients with both progressive disease and stable disease were eligible for enrollment, outcomes were similar in the two groups.
Ongoing studies with vandetanib include (1) an open-label phase II trial in patients under the age 18 years with familial MTC (with partial responses described in several patients including those with aggressive tumors associated with germ-line M918T RET mutations)73 and (2) a randomized placebo-controlled phase II trial in patients with metastatic DTC.
Potential synergistic combinations of vandetanib with other agents have also been of interest. Given the clinical evidence of vandetanib's efficacy in MTC, and in vitro evidence that bortezomib triggered caspase-dependent apoptosis in MTC cells, a phase I/II trial of the combination has been initiated, with enrollment targeting patients with advanced MTC as well as other solid tumors.74
Axitinib
Axitinib (AG-013736) is an oral inhibitor that effectively blocks VEGF receptors at subnanomolar concentrations, but notably not the RET kinase.75 In a phase I study of 36 patients with advanced solid malignancies, one of five thyroid cancer patients experienced tumor shrinkage, although none qualified as a partial response.76 A multicenter, open-label phase II study examined the efficacy of axitinib in advanced or metastatic thyroid carcinoma, starting at a dose of 5 mg twice daily.77 Of the 60 patients who started therapy, 50% had PTC, 25% had FTC (including Hurthle cell variants), and 18% had MTC. Although response assessment was not possible in 25% of the patients, confirmed partial response rate was 30% by intent-to-treat analysis (31% in DTC; 18% in MTC; 1 patient with ATC). Responses were observed in patients despite previous treatments with a variety of chemotherapeutic regimens. Median PFS was 18 months. Common adverse events included fatigue, stomatitis, proteinuria, diarrhea, hypertension, and nausea. Exploratory analyses of soluble biomarkers showed increases in serum VEGF levels, a recognized phenomenon of effective angiogenesis inhibition.78 Given the absence of inhibitory activity against RET or other mutated kinases that are oncogenic in thyroid carcinoma, the efficacy of axitinib suggests that VEGFR-mediated angiogenesis is likely the primary mechanism by which the other anti-VEGFR inhibitory agents function.
Currently ongoing is a multicenter, open-label phase II study to determine the efficacy of axitinib in patients with metastatic DTC refractory to doxorubicin, or for whom doxorubicin therapy is contraindicated.
Pazopanib
Pazopanib is a potent small-molecule inhibitor of all VEGFR subtypes as well as PDGFR. Like axitinib, it has insignificant significant inhibitory activity against the oncogenic kinases RET, RET/PTC and or BRAF, and therefore its actions are expected to be primarily antiangiogenic in thyroid carcinoma. Preliminary results were recently reported for 37 patients with rapidly progressing DTC treated in a phase II trial.79 With a starting daily dose of 800 mg, 32% of patients had confirmed partial responses, and the 6-month PFS was 71%. The most common side effects of therapy included hypertension in nearly half of the patients, elevated serum transaminases, headache, and mucositis. Preliminary results were also recently reported for 14 patients with rapidly progressing MTC treated in a phase II trial.79 One patient (7%) experienced a partial response; at the time of the report, eight (57%) were alive without progression, two (14%) were alive with progression, and four (29%) had died. The study has continued on to a second stage, enrolling up to 28 patients.
XL184
XL184 is an oral, small-molecule inhibitor of VEGF receptors 1 and 2, C-MET, RET, C-KIT, FLT3, and Tie-2.80 The inhibitory activity against C-MET, the cognate receptor for the hepatocyte growth factor, may provide additional synergistic benefit in thyroid carcinomas, given the enhanced expression of the receptor observed in PTC and MTC.81, 82, 83 An ongoing phase I dose-escalation study has examined the safety and pharmacokinetics of XL184 in patients with metastatic solid malignancies, with an expansion cohort limited to MTC.84 A total of 15 MTC patients (44%) had achieved at least 30% reduction in tumor measurements, with 10 (29%) having confirmed partial responses. No correlation was observed between RET mutation status (either germ line or somatic) and tumor response. A phase III trial, comparing XL184 with placebo, is now underway for patients with progressive, metastatic MTC.
Summary
The successful development of targeted therapies for cancer requires several key factors: (1) identification of biologically validated targets critical to development and maintenance of the malignant phenotype; (2) development of potent inhibitors of the targets, with broad therapeutic index separating efficacy from toxicity; (3) recognition of patient and tumor characteristics that can optimize the selection of patients for therapy; (4) identification of biomarkers predictive of patient outcome and that permit optimization of drug dosing; and (5) recognition of opportunities for well-tolerated and more efficacious combinatorial treatments. As summarized in this review, such advances have been made in the past few years in the development of successful targeted therapies for thyroid cancers.
Compared with the dismal historical track record, the recent proliferation of clinical trials for thyroid cancer has been remarkable. Targeting angiogenesis (and specifically VEGF receptors) has produced the most impressive clinical responses to date in both DTC and MTC. Although most small-molecule VEGF receptor antagonists also inhibit RET, the efficacy of axitinib and pazopanib to induce objective responses in the absence of any significant anti-RET activity suggests that RET may not be as important a target for therapy as VEGFR. Unfortunately, eventual progression despite antiangiogenic VEGFR blockade suggests emergence of alternate pathways to promote tumor growth and metastasis (including FGFR, C-MET, and angiopoietins).85 Further studies are necessary to explore the value of effective inhibition of the MAPK pathway downstream from oncogenic mutations, as well as other pathways stimulating tumor growth and metabolism such as PI3K-AKT-mTOR signaling.
The overall goal of developing new treatments is to extend the duration of life without unduly harming the quality of that life. Presently, no novel treatment has yet shown improved survival for thyroid cancer patients. The experience with other malignancies treated with VEGFR inhibitors suggests that survival may be improved by only a few months despite these radiographic tumor responses, and the possibility that short-term responses may be achieved at the cost of promoting greater tumor invasiveness and further metastases should be sobering.86 Toxicities of many of these new therapies, although less life-threatening than cytotoxic chemotherapies, are common and can be dose limiting, and clinicians must be familiar with recognizing and managing the side effects if they intend to use these agents. Finally, the low rate of partial response, the absence of complete responses, and emergence of resistance in all of the various monotherapy trials identify the need to develop either more effective single agents or to identify rational combinations of therapeutic targets (including cytotoxic chemotherapies) that have synergistic effectiveness without enhanced cross-toxicities.
References
Kilfoy BA, Zheng T, Holford TR, et al. International patterns and trends in thyroid cancer incidence, 1973–2002. Cancer Causes Control 2009;20:525–531.
Altekruse SF, Kosary CL, Krapcho M, et al (eds). SEER Cancer Statistics Review, 1975–2007. National Cancer Institute: Bethesda, MD, 2010.
Gilliland FD, Hunt WC, Morris DM, et al. Prognostic factors for thyroid carcinoma: a population-based study of 15,698 cases from the Surveillance, Epidemiology and End Results (SEER) program 1973–1991. Cancer 1997;79:564–573.
Smith BD, Smith GL, Hurria A, et al. Future of cancer incidence in the United States: burdens upon an aging, changing nation. J Clin Oncol 2009;27:2758–2765.
Sherman SI . Thyroid carcinoma. Lancet 2003;361:501–511.
Radio-iodine halts one type of cancer. Life 1949;54–56.
Trunnell JB . The treatment of human thyroid disease with radioactive iodine. Trans NY Acad Sci 1949;11:195–201.
Maloof F, Vickery AL, Rapp B . An evaluation of various factors influencing the treatment of metastatic thyroid carcinoma with I 131. J Clin Endocrinol Metab 1956;16:1–27.
Ronga G, Filesi M, Montesano T, et al. Lung metastases from differentiated thyroid carcinoma. A 40 years' experience. Q J Nucl Med Mol Imaging 2004;48:12–19.
Kondo T, Ezzat S, Asa SL . Pathogenetic mechanisms in thyroid follicular-cell neoplasia. Nat Rev Cancer 2006;6:292–306.
Roman S, Lin R, Sosa JA . Prognosis of medullary thyroid carcinoma: demographic, clinical, and pathologic predictors of survival in 1252 cases. Cancer 2006;107:2134–2142.
Sherman SI . Cytotoxic chemotherapy for differentiated thyroid carcinoma. Clin Oncol (R Coll Radiol) 2010;22:464–468.
Therasse P, Arbuck S, Eisenhauer E, et al. New guidelines to evaluate the response to treatment in solid tumors. European Organization for Research and Treatment of Cancer, National Cancer Institute of the United States, National Cancer Institute of Canada. J Natl Cancer Inst 2000;92:205–216.
Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer 2009;45:228–247.
Fagin JA . How thyroid tumors start and why it matters: kinase mutants as targets for solid cancer pharmacotherapy. J Endocrinol 2004;183:249–256.
Durante C, Puxeddu E, Ferretti E, et al. BRAF mutations in papillary thyroid carcinomas inhibit genes involved in iodine metabolism. J Clin Endocrinol Metab 2007;92:2840–2843.
Elisei R, Ugolini C, Viola D, et al. BRAF(V600E) mutation and outcome of patients with papillary thyroid carcinoma: a 15-year median follow-up study. J Clin Endocrinol Metab 2008;93:3943–3949.
Xing M, Clark D, Guan H, et al. BRAF mutation testing of thyroid fine-needle aspiration biopsy specimens for preoperative risk stratification in papillary thyroid cancer. J Clin Oncol 2009;27:2977–2982.
Carmeliet P . Mechanisms of angiogenesis and arteriogenesis. Nat Med 2000;6:389–395.
Ferrara N, Kerbel RS . Angiogenesis as a therapeutic target. Nature 2005;438:967–974.
Klein M, Vignaud JM, Hennequin V, et al. Increased expression of the vascular endothelial growth factor is a pejorative prognosis marker in papillary thyroid carcinoma. J Clin Endocrinol Metab 2001;86:656–658.
Lennard CM, Patel A, Wilson J, et al. Intensity of vascular endothelial growth factor expression is associated with increased risk of recurrence and decreased disease-free survival in papillary thyroid cancer. Surgery 2001;129:552–558.
Jo YS, Li S, Song JH, et al. Influence of the BRAF V600E mutation on expression of vascular endothelial growth factor in papillary thyroid cancer. J Clin Endocrinol Metab 2006;91:3667–3670.
Pfister DG, Fagin JA . Refractory thyroid cancer: a paradigm shift in treatment is not far off. J Clin Oncol 2008;26:4701–4704.
Knauf JA, Fagin JA . Role of MAPK pathway oncoproteins in thyroid cancer pathogenesis and as drug targets. Curr Opin Cell Biol 2009;21:296–303.
Santarpia L, Ye L, Gagel RF . Beyond RET: potential therapeutic approaches for advanced and metastatic medullary thyroid carcinoma. J Intern Med 2009;266:99–113.
Sleijfer S, Wiemer E, Verweij J . Drug insight: gastrointestinal stromal tumors (GIST)—the solid tumor model for cancer-specific treatment. Nat Clin Pract Oncol 2008;5:102–111.
Sala E, Mologni L, Truffa S, et al. BRAF silencing by short hairpin RNA or chemical blockade by PLX4032 leads to different responses in melanoma and thyroid carcinoma cells. Mol Cancer Res 2008;6:751–759.
Flaherty K, Puzanov I, Sosman J, et al. Phase I study of PLX4032: Proof of concept for V600E BRAF mutation as a therapeutic target in human cancer. J Clin Oncol (Meeting Abstracts) 2009;27 (Suppl):9000.
Salerno P, De Falco V, Tamburrino A, et al. Cytostatic activity of adenosine triphosphate-competitive kinase inhibitors in BRAF mutant thyroid carcinoma cells. J Clin Endocrinol Metab 2010;95:450–455.
Schwartz GK, Robertson S, Shen A, et al. A phase I study of XL281, a selective oral RAF kinase inhibitor, in patients (Pts) with advanced solid tumors. J Clin Oncol (Meeting Abstracts) 2009;27 (Suppl):3513.
Polverino A, Coxon A, Starnes C, et al. AMG 706, an oral, multikinase inhibitor that selectively targets vascular endothelial growth factor, platelet-derived growth factor, and kit receptors, potently inhibits angiogenesis and induces regression in tumor xenografts. Cancer Res 2006;66:8715–8721.
Coxon A, Bready J, Fiorino M, et al. Anti-tumor activity of AMG 706, an oral multi-kinase inhibitor, in human medullary thyroid carcinoma xenografts. Thyroid 2006;16:920.
Rosen LS, Kurzrock R, Mulay M, et al. Safety, pharmacokinetics, and efficacy of AMG 706, an oral multikinase inhibitor, in patients with advanced solid tumors. J Clin Oncol 2007;25:2369–2376.
Sherman SI, Wirth LJ, Droz JP, et al. Motesanib diphosphate in progressive differentiated thyroid cancer. N Engl J Med 2008;359:31–42.
Schlumberger M, Elisei R, Bastholt L, et al. Phase II study of safety and efficacy of motesanib (AMG 706) in patients with progressive or symptomatic, advanced or metastatic medullary thyroid cancer. J Clin Oncol 2009;27:3794–3801.
Xing M . BRAF mutation in papillary thyroid cancer: pathogenic role, molecular bases, and clinical implications. Endocr Rev 2007;28:742–762.
Bass MB, Davis MT, Kivman L, et al. Placental growth factor as a marker of therapeutic response to treatment with motesanib in patients with progressive advanced thyroid cancer, advanced nonsquamous non-small cell lung cancer, and locally recurrent or advanced metastatic breast cancer. J Clin Oncol (Meeting Abstracts) 2010;28 (Suppl):3037.
Sherman SI, Schlumberger MJ, Elisei R, et al. Exacerbation of postsurgical hypothyroidism during treatment of thyroid carcinoma with motesanib diphosphate (AMG 706). 89th Annual Meeting of The Endocrine Society: Toronto, ON, 2007.
Wilhelm SM, Carter C, Tang L, et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res 2004;64:7099–7109.
Carlomagno F, Anaganti S, Guida T, et al. BAY 43-9006 inhibition of oncogenic RET mutants. J Natl Cancer Inst 2006;98:326–334.
Strumberg D, Clark JW, Awada A, et al. Safety, pharmacokinetics, and preliminary antitumor activity of sorafenib: a review of four phase I trials in patients with advanced refractory solid tumors. Oncologist 2007;12:426–437.
Dubauskas Z, Kunishige J, Prieto V, et al. Cutaneous squamous cell carcinoma and inflammation of actinic keratoses associated with sorafenib. Clin Genitourin Cancer 2009;7:20–23.
Ratain MJ, Eisen T, Stadler WM, et al. Phase II placebo-controlled randomized discontinuation trial of sorafenib in patients with metastatic renal cell carcinoma. J Clin Oncol 2006;24:2505–2512.
Kloos RT, Ringel MD, Knopp MV, et al. Phase II trial of sorafenib in metastatic thyroid cancer. J Clin Oncol 2009;27:1675–1684.
Gupta-Abramson V, Troxel AB, Nellore A, et al. Phase II trial of sorafenib in advanced thyroid cancer. J Clin Oncol 2008;26:4714–4719.
Brose MS, Troxel AB, Redlinger M, et al. Effect of BRAFV600E on response to sorafenib in advanced thyroid cancer patients. J Clin Oncol (Meeting Abstracts) 2009;27 (Suppl):6002.
Hoftijzer H, Heemstra KA, Morreau H, et al. Beneficial effects of sorafenib on tumor progression, but not on radioiodine uptake, in patients with differentiated thyroid carcinoma. Eur J Endocrinol 2009;161:923–931.
Cabanillas ME, Waguespack SG, Bronstein Y, et al. Treatment with tyrosine kinase inhibitors for patients with differentiated thyroid cancer: the M.D. Anderson experience. J Clin Endocrinol Metab 2010;95:2588–2595.
Cabanillas ME, Kurzrock R, Sherman SI, et al. Phase I trial of combination sorafenib and tipifarnib: the experience in advanced differentiated thyroid cancer (DTC) and medullary thyroid cancer (MTC). J Clin Oncol (Meeting Abstracts) 2010;28 (Suppl):5586.
Ball DW . Medullary thyroid cancer: therapeutic targets and molecular markers. Curr Opin Oncol 2007;19:18–23.
Kober F, Hermann M, Handler A, et al. Effect of sorafenib in symptomatic metastatic medullary thyroid cancer. J Clin Oncol 2007;25 (Suppl):14065.
Lam ET, Ringel MD, Kloos RT, et al. Phase II clinical trial of sorafenib in metastatic medullary thyroid cancer. J Clin Oncol 2010;28:2323–2330.
Salvatore G, De Falco V, Salerno P, et al. BRAF is a therapeutic target in aggressive thyroid carcinoma. Clin Cancer Res 2006;12:1623–1629.
Nagaiah G, Fu P, Wasman JK, et al. Phase II trial of sorafenib (BAY 43-9006) in patients with advanced anaplastic carcinoma of the thyroid (ATC). J Clin Oncol 2009;27 (Suppl): abstract 6058.
Sherman SI . NCCN practice guidelines for thyroid cancer, version 2009a. 2009 [cited 1 June 2009]; available fromwww.nccn.org.
Waguespack SG, Sherman SI, Williams MD, et al. The successful use of sorafenib to treat pediatric papillary thyroid carcinoma. Thyroid 2009;19:407–412.
Kim DW, Jo YS, Jung HS, et al. An orally administered multitarget tyrosine kinase inhibitor, SU11248, is a novel potent inhibitor of thyroid oncogenic RET/papillary thyroid cancer kinases. J Clin Endocrinol Metab 2006;91:4070–4076.
Kelleher FC, McDermott R . Response to sunitinib in medullary thyroid cancer. Ann Intern Med 2008;148:567.
Dawson SJ, Conus NM, Toner GC, et al. Sustained clinical responses to tyrosine kinase inhibitor sunitinib in thyroid carcinoma. Anticancer Drugs 2008;19:547–552.
Cohen EEW, Needles BM, Cullen KJ, et al. Phase 2 study of sunitinib in refractory thyroid cancer. J Clin Oncol (Meeting Abstracts) 2008;26:6025.
Ravaud A, de la Fouchardière C, Courbon F, et al. Sunitinib in patients with refractory advanced thyroid cancer: the THYSU phase II trial. 2008, [cited 15 December 2008]; available fromhttp://www.asco.org/ASCO/Abstracts+%26+Virtual+Meeting/Abstracts?&vmview=abst_detail_view&confID=55&abstractID=33144.
Carr L, Goulart B, Martins R, et al. Phase II trial of continuous dosing of sunitinib in advanced, FDG-PET avid, medullary thyroid carcinoma (MTC) and well-differentiated thyroid cancer (WDTC). J Clin Oncol (Meeting Abstracts) 2009;27 (Suppl):6056.
Herbst RS, Heymach JV, O'Reilly MS, et al. Vandetanib (ZD6474): an orally available receptor tyrosine kinase inhibitor that selectively targets pathways critical for tumor growth and angiogenesis. Expert Opin Investig Drugs 2007;16:239–249.
Wedge SR, Ogilvie DJ, Dukes M, et al. ZD6474 inhibits vascular endothelial growth factor signaling, angiogenesis, and tumor growth following oral administration. Cancer Res 2002;62:4645–4655.
Carlomagno F, Vitagliano D, Guida T, et al. ZD6474, an orally available inhibitor of KDR tyrosine kinase activity, efficiently blocks oncogenic RET kinases. Cancer Res 2002;62:7284–7290.
Carlomagno F, Guida T, Anaganti S, et al. Disease associated mutations at valine 804 in the RET receptor tyrosine kinase confer resistance to selective kinase inhibitors. Oncogene 2004;23:6056–6063.
Holden SN, Eckhardt SG, Basser R, et al. Clinical evaluation of ZD6474, an orally active inhibitor of VEGF and EGF receptor signaling, in patients with solid, malignant tumors. Ann Oncol 2005;16:1391–1397.
Wells Jr SA, Gosnell JE, Gagel RF, et al. Vandetanib for the treatment of patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Oncol 2010;28:767–772.
Akeno-Stuart N, Croyle M, Knauf JA, et al. The RET kinase inhibitor NVP-AST487 blocks growth and calcitonin gene expression through distinct mechanisms in medullary thyroid cancer cells. Cancer Res 2007;67:6956–6964.
Robinson BG, Paz-Ares L, Krebs A, et al. Vandetanib (100 mg) in patients with locally advanced or metastatic hereditary medullary thyroid cancer. J Clin Endocrinol Metab 2010;95:2664–2671.
Wells SA, Robinson BG, Gagel RF, et al. Vandetanib (VAN) in locally advanced or metastatic medullary thyroid cancer (MTC): a randomized, double-blind phase III trial (ZETA). J Clin Oncol (Meeting Abstracts) 2010;28 (Suppl):5503.
Fox E, Widemann BC, Whitcomb PO, et al. Phase I/II trial of vandetanib in children and adolescents with hereditary medullary thyroid carcinoma. J Clin Oncol 2009;27 (Suppl): abstract 10014.
Mitsiades CS, McMillin D, Kotoula V, et al. Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. J Clin Endocrinol Metab 2006;91:4013–4021.
Inai T, Mancuso M, Hashizume H, et al. Inhibition of vascular endothelial growth factor (VEGF) signaling in cancer causes loss of endothelial fenestrations, regression of tumor vessels, and appearance of basement membrane ghosts. Am J Pathol 2004;165:35–52.
Rugo HS, Herbst RS, Liu G, et al. Phase I trial of the oral antiangiogenesis agent AG-013736 in patients with advanced solid tumors: pharmacokinetic and clinical results. J Clin Oncol 2005;23:5474–5483.
Cohen EE, Rosen LS, Vokes EE, et al. Axitinib is an active treatment for all histologic subtypes of advanced thyroid cancer: results from a phase II study. J Clin Oncol 2008;26:4708–4713.
Bocci G, Man S, Green SK, et al. Increased plasma vascular endothelial growth factor (VEGF) as a surrogate marker for optimal therapeutic dosing of VEGF receptor-2 monoclonal antibodies. Cancer Res 2004;64:6616–6625.
Bible KC, Smallridge RC, Maples WJ, et al. Phase II trial of pazopanib in progressive, metastatic, iodine-insensitive differentiated thyroid cancers. J Clin Oncol (Meeting Abstracts) 2009;27 (Suppl):3521.
Cui JJ . Inhibitors targeting hepatocyte growth factor receptor and their potential therapeutic applications. Expert Opin Ther Pat 2007;17:1035–1045.
Mineo R, Costantino A, Frasca F, et al. Activation of the hepatocyte growth factor (HGF)-Met system in papillary thyroid cancer: biological effects of HGF in thyroid cancer cells depend on Met expression levels. Endocrinology 2004;145:4355–4365.
Wasenius VM, Hemmer S, Karjalainen-Lindsberg ML, et al. MET receptor tyrosine kinase sequence alterations in differentiated thyroid carcinoma. Am J Surg Pathol 2005;29:544–549.
Papotti M, Olivero M, Volante M, et al. Expression of hepatocyte growth factor (HGF) and its receptor (MET) in medullary carcinoma of the thyroid. Endocr Pathol 2000;11:19–30.
Kurzrock R, Cohen EE, Sherman SI, et al. Long-term results in a cohort of medullary thyroid cancer (MTC) patients (pts) in a phase I study of XL184 (BMS 907351), an oral inhibitor of MET, VEGFR2, and RET. J Clin Oncol (Meeting Abstracts) 2010;28 (Suppl):5502.
Bergers G, Hanahan D . Modes of resistance to anti-angiogenic therapy. Nat Rev Cancer 2008;8:592–603.
Loges S, Mazzone M, Hohensinner P, et al. Silencing or fueling metastasis with VEGF inhibitors: antiangiogenesis revisited. Cancer Cell 2009;15:167–170.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
Research support: Genzyme, Eisai, AstraZeneca, Amgen, V Foundation for Cancer Research, National Cancer Institute. Consulting: Bayer, Exelixis, Plexxikon, Genzyme, Semafore, Eli Lilly, Oxigene, Celgene, Veracyte.
Honoraria: Genzyme, Exelixis.
Rights and permissions
About this article
Cite this article
Sherman, S. Targeted therapies for thyroid tumors. Mod Pathol 24 (Suppl 2), S44–S52 (2011). https://doi.org/10.1038/modpathol.2010.165
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/modpathol.2010.165
Keywords
This article is cited by
-
A comprehensive overview of the role of the RET proto-oncogene in thyroid carcinoma
Nature Reviews Endocrinology (2016)
-
Expression of Receptors for Pituitary-Type Growth Hormone-Releasing Hormone (pGHRH-R) in Human Papillary Thyroid Cancer Cells: Effects of GHRH Antagonists on Matrix Metalloproteinase-2
Hormones and Cancer (2015)
-
Modulation of Sodium Iodide Symporter in Thyroid Cancer
Hormones and Cancer (2014)
-
Evaluation of expression and function of vascular endothelial growth factor receptor 2, platelet derived growth factor receptors-alpha and -beta, KIT, and RET in canine apocrine gland anal sac adenocarcinoma and thyroid carcinoma
BMC Veterinary Research (2012)
-
Advances in the follow-up of differentiated or medullary thyroid cancer
Nature Reviews Endocrinology (2012)
