
Abstract
Preservation of macromolecules (DNA, RNA, and proteins) in tissue is traditionally achieved by immediate freezing of the sample. Although isolation of PCR-able RNA has been reported from formalin-fixed, paraffin-embedded tissues, the process has not been shown to be reproducible because high molecular weight RNA is usually degraded. We investigated the potential value of a new universal molecular fixative (UMFIX, Sakura Finetek USA, Inc., Torrance, California) in preservation of macromolecules in paraffin-embedded tissue. Mouse and human tissues were fixed in UMFIX from 1 hour to 8 weeks. They were then processed by a rapid tissue processing (RTP) system, embedded in paraffin, and evaluated for routine histology as well as for the quality and quantity of DNA, RNA, and proteins. Formalin-fixed tissues were processed by RTP and evaluated in a similar manner. Fresh-frozen samples were used as controls. The morphology of UMFIX-exposed tissue was comparable to that fixed in formalin. High molecular weight RNA was preserved in tissue that was immediately fixed in UMFIX and stored from 1 hour to 8 weeks at room temperature. There were no significant differences between UMFIX-exposed and frozen tissues on PCR, RT-PCR, real-time PCR, and expression microarrays. Similarly, physical and antigenic preservation of proteins in UMFIX tissue was similar to fresh state. Both RNA and proteins were substantially degraded in formalin-fixed and similarly processed specimens. We concluded that it is now possible to preserve histomorphology and intact macromolecules in the same archival paraffin-embedded tissue through the use of a novel fixative and a rapid processing system.
Similar content being viewed by others

Introduction
Progress in the understanding of molecular events that are involved in the development and progression of human disease is revolutionizing the way diseases are diagnosed and treated. Physicians and scientists are now harnessing the power of molecular techniques to diagnose and prognosticate pathologic disorders. Furthermore, it is now possible to direct therapeutic agents to specific products expressed by diseased cells without affecting normal tissues (Alizadeh et al, 2001; Bubendorf, 2001; Hillan and Quirke, 2001; Ross, 1999; Strausberg, 2001). On the other hand, while standard histopathologic methods maintain tissue architecture for morphologic assessment, they do not preserve macromolecules. The extraction of nucleic acids from formaldehyde-fixed, paraffin-embedded tissue, the most widely available material for clinical studies, is a notoriously unreliable and irreproducible process (Lewis et al, 2001). Therefore, macromolecules are usually extracted from fresh or snap-frozen tissues. Fresh or frozen tissue specimens, however, are of limited value for the assessment of histomorphology and cannot be used for long-term retrospective studies. Similarly, currently available tissue preservatives that protect nucleic acids cause considerable damage to the cell and tissue architecture and render them unsuitable for histomorphologic evaluation (Gillespie et al, 2002).
We herewith report the development of a simple and practical tissue fixative that preserves histomorphology and protects high quality DNA, RNA, and proteins at ambient temperature.
Results
DNA
The quality of restriction enzyme-digested DNA from frozen mouse liver tissue and mouse liver tissue treated with universal molecular fixative (UMFIX, Sakura Finetek USA, Inc., Torrance, California) were similar (Fig. 1). PCR amplification of DNA extracted from fresh, UMFIX-, and formalin-exposed tissues (Fig. 2A) also showed comparable size bands (~450 bp for G3PDH). However, in real-time PCR, a higher crossing threshold for formalin-exposed specimens was observed when compared with UMFIX samples (Fig. 2B). Threshold increased with longer tissue exposure to formalin. The quality and quantity of DNA in human tissue was not significantly affected by the time interval between surgical excision and immersion of tissue sections in either fixative.

Agarose gel electrophoresis of DNA isolated from fresh mouse liver (Lanes 1, 4, and 7) exposed to UMFIX (universal molecular fixative) for 1 hour (Lanes 2, 5, and 8) and 24 hours (Lanes 3, 6, and 9). L denotes 1000-bp ladder. Ten micrograms of the DNA was digested with 5 U/μg restriction enzymes, TaqI (at 65° C) (Lanes 1 to 3), BamHI (Lanes 4 to 6), and ECO-RI (at 37° C) (Lanes 7 to 9) overnight.

(A) Result of (Agilent DNA chip) PCR for G3PDH of DNA extracted from fresh mouse liver (Lane 3), exposed to UMFIX (universal molecular fixative) for 1 hour (Lane 4) and 24 hours (Lane 5), and formalin for 1 hour (Lane 6) and 24 hours (Lane 7). Lane 1, no template; Lane 2, positive control. (B) Result of quantitative PCR for G3PDH of DNA extracted from fresh mouse liver (crossing threshold [CT] = 14.48), exposed to UMFIX for 1 hour (CT = 14.21) and 24 hours (CT = 13.05), and formalin for 1 hour (CT = 16.14) and 24 hours (CT = 0). No template (CT = 0), positive control (CT = 10.67).
RNA
The quality and quantity of mouse liver RNA extracted from the frozen and UMFIX-exposed tissue was comparable, whereas formalin-fixed tissues yielded significantly degraded RNA, as indicated by the absence of 28S and 18S ribosomal RNA bands (Fig. 3). Although the RNA extracted from formalin-fixed tissue was significantly degraded, it could be used to amplify small amplicons by RT-PCR. The threshold crossing of the formalin-fixed tissue was significantly higher, however, compared with fresh and UMFIX-exposed samples (Fig. 4). Similar to DNA, threshold crossing was unchanged by longer exposure to UMFIX but was significantly different for formalin.

Agarose gel electrophoresis of total RNA isolated from mouse liver tissue. Fresh (Lane 1), exposed to UMFIX (universal molecular fixative) for 1 hour (Lane 2) and 24 hours (Lane 3), and formalin for 1 hour (Lane 4) and 24 hours (Lane 5). Arrows indicate 28S and 18S ribosomal RNA.

Result of quantitative RT-PCR for G3PDH of total RNA extracted from fresh mouse liver (CT = 15.14), exposed to UMFIX (universal molecular fixative) for 1 hour (CT = 18) and 24 hours (CT = 18.21), and formalin for 1 hour (CT = 21.95) and 24 hours (CT = 27.62). No template (CT = 0), positive control (CT = 10.08).
UMFIX-exposed mouse liver tissue processed by the rapid processing method under controlled RNase-free conditions and embedded in paraffin yielded an intact RNA comparable to that of fresh tissue (Fig. 5). Furthermore, human tissues that were fixed in UMFIX, processed by rapid processing method, and embedded in paraffin yielded nondegraded RNA up to 8 weeks after storage at room temperature (Fig. 6A). The cDNA array profiles of the extracted RNA from fresh-frozen samples and UMFIX-treated paraffin-embedded tissues exhibited comparable patterns. The expression levels were highly similar at various time points postprocessing (Fig. 6B) up to 8 weeks (Pearson coefficient r > 0.85, p < 0.05). The minor difference between the results was attributed to tissue heterogeneity.

Evaluation of quality of total RNA isolated from paraffin block and control fresh mouse liver. Samples were fixed in UMFIX (universal molecular fixative) and processed by rapid tissue processing (RTP) method. (A) Agarose gel electrophoresis; arrows indicate 28S and 18S ribosomal RNA. (B) Same samples were run on Agilent RNA 6000 Nanochip with resulting 28S/18S ratios (top of each histogram).

(A) Agarose gel electrophoresis of total RNA isolated from paraffin block of a metastatic carcinoma of the brain. Samples were fixed in UMFIX (universal molecular fixative) and processed by rapid tissue processing method for 1 day (Lane 1), 4 weeks (Lane 2), and 8 weeks (Lane 3) postprocessing. Arrows indicate 28S and 18S ribosomal RNA. (B) Expression ratios of 94 apoptosis-related genes to cyclophilin. Total RNA isolated from 1 day (Lane 1), 4 weeks (Lane 2), and 8 weeks post-processing of paraffin block of the same samples as A. Colored expression ratio scale shown at bottom.
The quality and quantity of RNA from human tissue was adversely affected by time interval between surgical excision and immersion of sections in UMFIX. When tissues were collected in the operating room and a section was immediately fixed, the results were reproducible. On the other hand, tissues fixed in UMFIX from 5 to 30 minutes postexcision yielded RNA of unpredictable quality. Similarly, UMFIX-exposed tissues, which were processed without controlled RNase-free environment, yielded RNA of variable quality.
Protein
Mouse liver preserved in UMFIX revealed an identical pattern with frozen samples on 2D-gel (Fig. 7). In contrast to the distinct spots observed with UMFIX samples, protein extracts from formalin-fixed tissue produced a smear without any distinct spots. Furthermore, no similarity could be observed between fresh samples and formalin-exposed samples by PDQuest software (Bio-Rad, Hercules, California), despite easing the matching criteria. Evaluation of number and location of spots on the gels aided by PDQuest software showed considerable homology between UMFIX samples and protein extracts of the fresh tissue. The same findings were seen on 1D-gels; distinct bands were observed with fresh and UMFIX samples, whereas formalin-fixed material produced a nondistinctive smear. Similarly, Western blots from 1D-gels showed strong distinct bands for UMFIX samples compared with formalin-treated material. Tested antibodies were chosen based on their molecular weight and cellular location. All of the 10 tested and 12 control antibodies reacted with samples fixed for 1 hour in UMFIX; twenty of those antibodies also had strong detectable bands in samples fixed up to 24 hours. Conversely, only six antibodies were reactive in samples fixed in formalin, with a considerable lower intensity (Fig. 8 and Table 1). The time interval between surgical excision and tissue fixation (usually < 30 minutes) did not affect the quality of extracted proteins.

Two-dimensional protein gel electrophoresis of mouse liver tissue in fresh state (A) and after 1-hour incubation with UMFIX (universal molecular fixative) (B) or formalin (C).

Western blot of mouse liver protein extracts in fresh state (A) and following exposure to UMFIX (universal molecular fixative) for 1 hour (B) and 24 hours (C), or formalin for 1 hour (D) and 24 hours (E). Lane 1: caveolin 1 (22 kD), Lane 2: casein kinase II alpha (45 kD), Lane 3: β-catenin (92 kD), Lane 4: Bcl-2 (26 kD), Lane 5: adaptin beta (106 kD), Lanes 6 and 7: control antibodies (see Table 1), Lane 8: GDNFR-alpha (45 to 60 kD), Lane 9: PKC alpha (82kD), Lane 10: PAF 53 (53 kD), Lane 11: PLC gamma (148 kD), and Lane 12: NF-KB p65 (65kD). MW = molecular weight in kilodalton (kD).
Histomorphology of Tissue
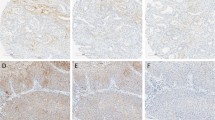
Hematoxylin and eosin–stained slides of UMFIX and formalin-fixed tissue showed no significant differences in tissue architecture, cellular and nuclear morphology, or tinctorial reaction (Fig. 9, A and B). The only exception was a moderate swelling of erythrocytes in samples exposed to UMFIX for more than 48 hours.

Histology, histochemistry, and immunohistochemistry of UMFIX (universal molecular fixative)-exposed tissue. (A) Hematoxylin and eosin–stained section of metastatic carcinoma of the brain (×400). (B) Hematoxylin and eosin–stained section of normal endometrium (×400). (C) Cirrhosis of the liver, Masson’s Trichrome (×200). (D) Mucicarmine staining of metastatic colonic carcinoma of the liver (×200). Immunoreactivity for (E) estrogen receptor and (F) desmin in normal myometrium (×400). (G) HER-2 in breast cancer (×200). (H) cytokeratin 20 in metastatic colonic carcinoma (×200).
Histochemistry and Immunohistochemistry
Histochemical (Fig. 9, C and D) and immunohistochemical (Fig. 9, E to H) stains of UMFIX-preserved tissue were comparable to those seen in formalin-fixed and routinely processed samples. However, immunoreactivity for a number of antibodies was slightly stronger in UMFIX-preserved tissue. The only exception was a moderate reduction in the intensity of reaction for the hepatocellular antigen (Hep Par 1) in UMFIX-fixed material. The quality and quantity of nuclear antigens did not differ significantly between the tissues fixed in formalin and UMFIX.
Discussion
The time-honored formalin fixation and routine tissue processing methods are of limited, if any, value in preserving macromolecules (Sambrook et al, 2001). Therefore, a reassessment of the standard diagnostic tissue preparation is required to accommodate tissue-based molecular procedures (Lewis et al, 2001). Ideal tissue fixation and processing procedures should preserve histomorphologic features, similar to that seen in formalin-fixed paraffin-embedded material, while protecting DNA, RNA, and protein in a manner comparable to fresh-frozen tissue. This will enable the pathologist to render a histologic diagnosis and use the same tissue slide, or corresponding paraffin block, to isolate intact macromolecules, hence allowing retrospective molecular studies on archival histologic material.
RNA is a very difficult macromolecule to protect during handling of tissue. Although isolation of RNA from formalin-fixed paraffin-embedded tissue has been reported, the extracted RNA is significantly degraded and can only be used for RT-PCR with amplicon sizes smaller than 400 bp (Mizuno et al, 1998; Specht et al, 2001; Tyrrell et al, 1995). In fact, others have suggested that fragments longer than 200 bp could not be reproducibly amplified from the formalin-fixed material (Lewis et al, 2001). Aside from degradation of RNA by formalin, there is also structural alteration of template that makes amplification methods irreproducible (Masuda et al, 1999). Our study demonstrates that the ill effect of formalin on nucleic acid templates is also time dependent. In contrast to formaldehyde-fixed samples, amplification curves of UMFIX-treated samples were similar to fresh specimens, indicating preservation of intact templates.
Tissue fixation in formalin at 4° C may yield RNA of better quality; the process, however, is inconvenient for the routine laboratory use and has not gained wide acceptance (Noguchi et al, 1997). Addition of ethylenediamine-4N′-tetraacetic acid to formalin has also been shown to improve the quality of RNA for amplification of longer segments. The resulting RNA is nevertheless still significantly degraded (Noguchi et al, 1997). The discouraging results with formalin-fixed tissue have prompted investigators to seek alternative fixatives for molecular studies such as acetone- and alcohol- based solutions. Although superior to formalin with regard to RNA preservation, the requirement for fixation at 4° C or −20° C has rendered them impractical for routine laboratory use (Sato et al, 1991; Shibutani et al, 2000).
A high sulfate solution (RNAlater, Ambion Inc., Austin, Texas) that protects tissue RNA at 4° C and ambient temperature was recently reported (Florell et al, 2001). Following incubation in this solution, tissue can be postfixed in formalin and processed, resulting in histomorphology comparable to those seen with routine formalin fixation (Florell et al, 2001). Its major drawback is that it does not permit histologic diagnosis and extraction of macromolecules from the same slide or tissue block.
Formalin fixation not only damages RNA but also induces structural changes in proteins by cross-linking. Our data, on the other hand, show that on 1-dimensional and 2-dimensional gels, UMFIX-treated and fresh tissue produce highly similar patterns, suggesting that UMFIX induces limited or no alteration in protein structure. In contrast to formalin fixation, antigenic properties of proteins for the most part was unchanged in UMFIX-treated samples.
The present study shows that it is feasible to simultaneously protect histomorphology and the integrity of macromolecules in fixed and processed tissue. The UMFIX reagent used in our study seems to provide enormous advantage over the conventional fixation methods in allowing diagnosis, prognostication, and identification of treatment targets in patient samples. The fact that RNA is preserved in paraffin-embedded tissue following UMFIX exposure and rapid tissue processing (RTP) allows for performing microdissection and molecular profile of the same histologic section used for diagnosis. Preservation of proteins in these samples suggests that the same material could be used for proteomic studies.
The successful utilization of the above-described system in clinical settings requires a reassessment of current practice of handling human tissue from the operating room to the laboratory. Rapid fixation of tissue blocks immediately following surgical excision is the most important step in ensuring the quality of RNA. Furthermore, it is essential not to expose tissue to formalin, either during fixation or processing. The process also requires changes in the work habits of the laboratory personnel, including adaptation to an RNA-friendly environment. The benefits derived from the herein-described fixation and processing methodologies, however, will far outweigh inconveniences that may occur in the early stages of their implementation.
Materials and Methods
Preparation of Special Fixative
UMFIX (Sakura Finetek USA, Inc.) is a mixture of methanol and polyethylene glycol at a predetermined ratio as previously described (US patent application number 10/141, 780).
Tissue Samples
Both mouse and human samples were used in this study. In the initial phase, mouse liver tissue was used to determine the effect of UMFIX on macromolecules. Mouse liver was excised from 3-month-old C57BL/6 female mice. Cube-shaped tissue fragments weighing approximately 50 mg were then immediately immersed in UMFIX or 10% phosphate-buffered formalin. Similar sized cubes were snap-frozen in liquid nitrogen and stored at −75° C to serve as control specimens.
In the second phase of the study, human tissues were used both for molecular assays and determination of UMFIX effect on histomorphology as well as on histochemical and immunohistochemical properties. Human tissues (adrenal, breast, colon, eye, esophagus, kidney, liver, lung, lymph node, skeletal muscle, pancreas, parathyroid, parotid, prostate, skin, small intestine, soft tissue, spleen, thyroid, tonsil, and uterus) were collected from 1 to 30 minutes after surgery at the University of Miami, Jackson Memorial Medical Center. Representative samples were taken primarily from the large surgically excised specimens. The main surgical specimens were fixed in 10% neutral buffered formalin and processed overnight in a standard processing machine (Carson, 1997). Parallel tissue blocks were fixed in the UMFIX and 10% neutral buffered formalin and were processed according to a recently described rapid processing technique under controlled RNase-free conditions (Morales et al, 2002). Adjacent sections were also snap-frozen in liquid nitrogen. Mouse and human tissues were stored in UMFIX at both 4° C and ambient temperature (22° C) for periods of time ranging from 1 hour to 8 weeks.
DNA Extraction
Approximately 50 mg of frozen or UMFIX-preserved mouse liver tissue was used to extract DNA (Puregene Kit, Gentra, Minneapolis, Minneapolis). The extracted DNA was evaluated using agarose gel electrophoresis and spectrophotometry (Ultraspec III, Pharmacia, the Netherlands). Ten micrograms of the DNA was digested with 5 U/μg restriction enzymes, TaqI (65° C) and BamH I, and EcoR I (37° C) overnight. Following digestion, DNA was resolved on 1% agarose gel.
RNA Extraction
Total RNA was extracted from frozen, UMFIX-, and formalin-immersed tissue. RNA was also prepared from the same tissue after processing by RTP. Extraction was performed by addition of Trizol reagent (GibcoBRL, Gaithersburg, Maryland) and subsequent homogenization using a tissue tearor (Biospec Products Inc., Bartlesville, Oklahoma). The RNA from homogenized tissue was extracted with chloroform followed by isopropyl precipitation on ice. The RNA pellets were resuspended in 100 μl of diethylpyrocarbonate-treated water. Standard 1% agarose gel under denaturing condition with ethidium bromide was used to assess the integrity of RNA. In addition, RNA was run on Agilent Technologies Bioanalyzer 2100 using RNA 6000 Nano Chip (Lindenhurst, New York) to determine the RNA integrity and the ratio of ribosomal RNA. The quantity of the extracted RNA was determined by spectrophotometery (Ultraspec III, Pharmacia).
PCR, RT-PCR, and Real-Time PCR
PCR was performed using mouse G3PDH primers (Clonetech, Palo Alto, California) using 0.5 μg of Rnase-treated isolated DNA and Qiagen TaqPCR Mastermix (Qiagen, Valencia, California). Alternatively, the same primer was used with Qiagen Quantitect Sybrgreen Mastermix on Bio-Rad I-cycler. The conditions for DNA PCR were as follows: 95° C, 15 minutes; 35 cycles at 94° C, 45 seconds; 60° C, 45 seconds; 72° C, 2 minutes; and hold for 7 minutes at 72° C. Furthermore, mouse G3PDH primers (Biosource, Camarillo, California) were used with 0.5 μg of DNase-treated RNA for real-time RT-PCR using Qiagen Quantitect Sybrgreen Mastermix or Qiagen Quantitect probe Mastermix on Bio-Rad I-cycler. Conditions for PCR were an initial reverse transcription for 30 minutes at 50° C followed by Taq activation at 95° C for 20 minutes followed by 40 cycles at 95° C 15 seconds and 60° C for 1 minute.
Expression Profile
Total RNA was extracted from UMFIX-treated tissues before and after processing and paraffin embedding and used for nylon-based cDNA microarrays of apoptosis-related genes (HS-002–12, Superarray, Bethesda, Maryland) following manufacturer’s protocol. X-ray densitometric results were obtained by Chemimager 5500 Gel Documentation System (Alpha Innotech Corporation, San Leandro, California). Data were analyzed using Genesis software (Genesis, Graz, Austria). Statistical analysis was performed with the aid of Statistica software (StatSoft, Tulsa, Oklahoma).
Protein Studies
Total protein was extracted using a standard protocol (Sambrook et al, 2001). Briefly, samples of mouse liver were homogenized in 20 μl T-PER reagent per milligram of tissue (Pierce Biotechnology, Rockford, Illinois). Protein concentration was measured using Coomassie protein assay (Bio-Rad) using BSA as standard. For 2D-gel experiment, protein extract was used according to the ReadyPrep 2-D Starter Kit instructions (Bio-Rad). Briefly, 11-cm IPG (pH 3 to 10) strips were incubated overnight at room temperature with 250 μg of protein in 185 μl of rehydration buffer containing 10 ml of 8 M urea, 2% CHAPS, 50 mm dithiothreitol (0.2% w/v), Bio-Lyte 3/10 Ampholytes, and Bromophenol Blue. Rehydrated strips were then transferred to Immunoelectrophoresis Focusing Chamber (Bio-Rad) and run according to the manufacturer’s recommended protocol (5.3 hours at 30,000 volt/hr). Strips were then equilibrated for 10 minutes in a mixture of 6 M urea, 2% SDS, 0.375 M Tris-HCl (pH 8.8), 20% glycerol, and 2% (w/v) DTT, and then 10 minutes in the same buffer without DTT but with added iodoacetamide. The strips were then laid over the 11 cm criterion gels and subjected to electrophoresis in the Criterion System (Bio-Rad), at 200 V, constant for 65 minutes using 1X Tris/glycine/SDS running buffer. Gels were then washed in 10% methanol and 7% acetic acid for 30 minutes and stained overnight using Sypro-Ruby protein stain (Bio-Rad). After a 10-minute wash in 10% methanol and 7% acetic acid, gels were rinsed in ddH2O and analyzed using a Chemimager 5500 Gel Documentation System (Alpha Innotech Corporation) equipped with CCD camera. Tiff images were analyzed for automated spot matching using PDQuest software (Bio-Rad).
Alternatively, protein was isolated by homogenizing the tissue and adding 4.1 ml of boiling lysis buffer (10 mm Tris, pH 7.4, 1 mm sodium ortho-vanadate, 1% SDS) for 15 to 20 seconds. Next, 0.9 ml of ×6 electrophoresis sample buffer (360 mm Tris pH 6.8, 600 mm DTT, 12% SDS, 60% glycerol, 0.018% bromophenol blue) was added and mixed well. Samples were heated again at 95° C for 30 seconds in heat blocks.
Western blot transfer was done according to a standard protocol (performed by Beckton Dickinson Powerblot Service, Lexington, Kentucky). The antibodies used were selected based on their activity against proteins with different various cellular location and molecular weight. The antibodies were against caveolin 1, casein kinase II alpha, β-catenin, Bcl-2, adaptin beta, GDNFR-alpha, PKC alpha, PAF53, NF-kappa B (p65), and phospholipase C gamma (Beckton Dickinson Powerblot Service). In addition, another 13 antibodies were run as control (Table 1). Briefly, 66 μg of protein was loaded in each well across the entire width of the 4% to 15% gradient SDS-polyacrylamide gel (Bio-Rad Criterion IPG Well Comb). The gel was run for 1.5 hours at 150 volts then transferred to Immobilon-P membrane (Millipore, Bedford, Massachusetts) for 2 hours at 200 mAmp, using a Hoefer wet electrophoretic transfer apparatus, TE Series (Amersham Biosciences, Piscataway, New Jersey). Following transfer the membrane was immersed for 1 hour with gelatin blocking buffer. Next, the membrane was clamped with a Western blotting manifold that isolates 40 channels across the membrane. In each channel, a primary antibody mix was added and incubated for 1 hour at 37° C. The blot was removed from the manifold, washed, and incubated for 45 minutes at 37° C with secondary goat antimouse Ig conjugated to Alexa680 fluorescent dye (Molecular Probes, Eugene, Oregon). The membrane was washed, dried, and scanned using the Odyssey Infrared Imaging System (LI-COR, Lincoln, Nebraska).
Histology, Histochemistry, and Immunohistochemistry
Four-micron–thick sections of paraffin-embedded tissue processed by the conventional as well as the rapid technique were prepared for routine hematoxylin and eosin as well as histochemical and immunohistochemical stains (Carson, 1997; Morales et al, 2002). To evaluate histomorphology, special attention was paid to the overall pattern of tissue preservation, cellular and extra cellular structures, and the cell and nuclear morphology, as well as to tinctorial reactions of various tissue and cell components.
Periodic acid-Schiff with or without diastase digestion, Alcian blue at pH 2.5, Mayer’s mucicarmine, and Masson’s trichrome stains were done following standard protocols (Carson, 1997). Immunohistochemistry was performed using labeled streptavidin-biotin-peroxidase technique with antibodies to the following antigens: caldesmon, carcinoembryonic antigen, CD3, CD20, CD31, CD68, chromogranin, CK7, CK20, desmin, epithelial membrane antigen, estrogen receptor, factor VIII, Hep Par 1, HER2, high molecular weight keratin, kappa light chain, lambda light chain, leukocyte common antigen, p63, PLAP, progesterone receptor, prostatic specific antigen, S100 protein, trypsin, TTF-1 (DAKO Corporation, Carpinteria, California), Ki67 (Beckton Dickinson, Franklin Lakes, New Jersey), p53 (Oncogene Research Product, San Diego, California), and RET oncoprotein (Vector Laboratories, Burlingame, California). Whenever needed, antigen retrieval was carried out using citrate buffer in a vegetable steamer. Diaminobenzidine constituted the chromogenic substrate. For cytoplasmic antigens, a nuclear counterstain (hematoxylin) was used. For visualization of nuclear antigens, the diaminobenzidine color was first intensified by 1% cupric sulfate. The slides were then counterstained by fast green. The intensity, pattern, and specificity of the immunohistochemical reactions were assessed and compared on all slides.
All authors contributed equally to the efforts presented in this paper.
References
Alizadeh AA, Ross DT, Perou CM, and van de Rijn M (2001). Toward a novel classification of human malignancies based on gene expression patterns. J Pathol 195: 41–52.
Bubendorf L (2001). High-throughput microarray technologies: From genomics to clinics. Eur Urol 40: 231–238.
Carson FL (1997). Histotechnology: A Self-Instructional Text, 2nd ed. Chicago: ASCP Press.
Florell SR, Coffin CM, Holden JA, Zimmerman JW, Gerwels JW, Summers BK, Jones DA, and Leachman SA (2001). Preservation of RNA for functional genomic studies: A multidisciplinary tumor bank protocol. Mod Pathol 14: 116–128.
Gillespie JW, Best CJ, Bichsel VE, Cole KA, Greenhut SF, Hewitt SM, Ahram M, Gathright YB, Merino MJ, Strausberg RL, Epstein JL, Hamilton SR, Gannot G, Baibakova GV, Calvert VS, Flaig MJ, Chuaqui RF, Herring JC, Pfeifer J, Petricoin EF, Linehan WM, Duray PH, Bova GS, and Emmert-Buck MR (2002). Evaluation of non-formalin tissue fixation for molecular profiling studies. Am J Pathol 160: 449–457.
Hillan KJ and Quirke P (2001). Preface to genomic pathology: A new frontier. J Pathol 195: 1–2.
Lewis F, Maughan NJ, Smith V, Hillan K, and Quirke P (2001). Unlocking the archive gene expression in paraffin-embedded tissue. J Pathol 195: 66–71.
Masuda N, Ohnishi T, Kawamoto S, Monden M, and Okubo K (1999). Analysis of chemical modification of RNA from formalin-fixed samples and optimization of molecular biology applications for such samples. Nucleic Acids Res 27: 4436–4443.
Mizuno T, Nagamura H, Iwamoto KS, Ito T, Fukuhara T, Tokunaga M, Tokuoka S, Mabuchi K, and Seyama T (1998). RNA from decades-old archival tissue blocks for retrospective studies. Diag Mol Pathol 7: 202–208.
Morales AR, Essenfeld H, Essenfeld E, Duboue MC, Vincek V, and Nadji M (2002). Continous-specimen-flow, high-throughput, 1-hour tissue processing: A system for rapid diagnostic tissue preparation. Arch Pathol Lab Med 126: 583–590.
Noguchi M, Furuya S, Takeuchi T, and Hirohashi S (1997). Modified formalin and methanol fixation methods for molecular biological and morphological analyses. Pathol Int 47: 685–691.
Ross JS (1999). The impact of molecular diagnostic tests on patient outcomes. Clin Lab Med 19: 815–831.
Sambrook J, Russell DW, and Sambrook J (2001). Molecular Cloning. A Laboratory Manual, 3rd ed. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press.
Sato Y, Mukai K, Furaya S, and Shimosato Y (1991). The Amex method: A multipurpose tissue-processing and paraffin-embedding method. III. Extraction and purification of RNA and application to purification of RNA and application to slot-blot hybridization analysis. J Pathol 163: 81–85.
Shibutani M, Uneyama C, Miyazaki K, Toyoda K, and Hirose M (2000). Methacarn fixation: A novel tool for analysis of gene expression in paraffin-embedded tissue specimens. Lab Invest 80: 199–208.
Specht K, Richter T, Muller U, Walch A, Werner M, and Hofler H (2001). Quantitatiave gene expression analysis in microdissected archival formalin-fixed and paraffin-embedded tumor tissue. Am J Pathol 158: 419–429.
Strausberg RL (2001). The cancer genome anatomy project: New resources for reading the molecular signature of cancer. J Pathol 195: 31–40.
Tyrrell L, Elias J, and Longley J (1995). Detection of specific mRNA in routinely processed dermatopathology specimens. Am J Dermatopathol 17: 476–483.
Acknowledgements
Supported in part by Sakura Finetek USA, Inc., Torrance, California.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Vincek, V., Nassiri, M., Nadji, M. et al. A Tissue Fixative that Protects Macromolecules (DNA, RNA, and Protein) and Histomorphology in Clinical Samples. Lab Invest 83, 1427–1435 (2003). https://doi.org/10.1097/01.LAB.0000090154.55436.D1
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1097/01.LAB.0000090154.55436.D1
Keywords
This article is cited by
-
Coping with formalin banning in pathology: under vacuum long-term tissue storage with no added formalin
Histochemistry and Cell Biology (2019)
-
Protection of tissue physicochemical properties using polyfunctional crosslinkers
Nature Biotechnology (2019)
-
MassARRAY, pyrosequencing, and PNA clamping for EGFR mutation detection in lung cancer tissue and cytological samples: a multicenter study
Journal of Cancer Research and Clinical Oncology (2016)
-
A formalin-free method for stabilizing cells for nucleic acid amplification, hybridization and next-generation sequencing
BMC Research Notes (2015)
-
Quantitation of fixative-induced morphologic and antigenic variation in mouse and human breast cancers
Laboratory Investigation (2013)
