
Abstract
The human serum protein transthyretin (TTR) is highly fibrillogenic in vitro and is the fibril precursor in both autosomal dominant (familial amyloidotic polyneuropathy [FAP] and familial amyloidotic cardiomyopathy [FAC]) and sporadic (senile systemic amyloidosis [SSA]) forms of human cardiac amyloidosis. We have produced mouse strains transgenic for either wild-type or mutant (TTRLeu55Pro) human TTR genes. Eighty-four percent of C57Bl/6xDBA/2 mice older than 18 months, transgenic for the wild-type human TTR gene, develop TTR deposits that occur primarily in heart and kidney. In most of the animals, the deposits are nonfibrillar and non-Congophilic, but 20% of animals older than 18 months that bear the transgene have human TTR cardiac amyloid deposits identical to the lesions seen in SSA. Amino terminal amino acid sequence analysis and mass spectrometry of the major component extracted from amyloid and nonamyloid deposits revealed that both were intact human TTR monomers with no evidence of proteolysis or codeposition of murine TTR. This is the first instance in which the proteins from amyloid and nonfibrillar deposits in the same or syngeneic animals have been shown to be identical by sequence analysis. It is also the first time in any form of amyloidosis that nonfibrillar deposits have been shown to systematically occur temporally before the appearance of fibrils derived from the same precursor in the same tissues. These findings suggest, but do not prove, that the nonamyloid deposits represent a precursor of the fibril. The differences in the ultrastructure and binding properties of the deposits, despite the identical sizes and amino terminal amino acid sequences of the TTR and the dissociation of deposition and fibril formation, provide evidence that in vivo factors, perhaps associated with aging, impact on both systemic precursor deposition and amyloid fibril formation.
Similar content being viewed by others
Introduction
To date, 20 different proteins have been identified as the fibril precursor in human disease-related amyloidoses (Buxbaum and Tagoe, 2000). In each instance the deposits display Congo red binding with positive birefringence under polarization microscopy, a discrete fibrillar ultrastructure and the colocalization of serum amyloid P component (SAP), features which define the material as amyloid. The prevalence of some forms of tissue amyloid deposition—notably those derived from wild-type protein molecules: transthyretin, islet amyloid polypeptide (IAPP), atrial naturietic factor (ANF), apolipoprotein A1 (ApoA1), and Alzheimer β protein precursor (AβPP)—increases with age, a phenomenon noted even before the unique chemical nature of each of the individual precursors was established (Wright and Calkins, 1981).
Studies with many of the precursors have shown that aggregates with the ultrastructural appearance of fibrils and the tinctorial properties of in vivo amyloid can be produced in the test tube in the absence of any additional macromolecules. These observations have justifiably been interpreted as indicating that fibrillogenesis is an intrinsic property of the precursor protein (Kelly and Lansbury, 1994). The pathway from the soluble state to deposited fibril is not fully understood for any precursor. In the case of transthyretin, it appears that a misfolded monomer populates the protofibril pool. However, it is uncertain if aggregates with varying degrees of solubility precede protofibril and fibril formation.
In patients, even the most amyloidogenic precursors, which produce early onset familial autosomal dominant disease, do not form fibrillar deposits until mid to late adulthood. Based on data obtained with the Aβ protein, the temporal delay has been attributed to an ongoing slow nucleation reaction in which the rate of fibril formation increases with the mass of fibrils available. It is more likely that in vivo the process may be modified by factors that prevent fibril deposition early in life. These unknown elements may become less effective with increasing age. Alternatively, mechanisms that promote amyloidogenesis may become more active with advancing years. In either case, it seems likely that the fibrillogenic potential of any precursor, as demonstrated in vitro, requires additional elements to achieve full expression in vivo.
Normal sequence human serum protein transthyretin (TTR) amyloid deposits are present in the ventricular myocardium of as many as 25% of autopsied individuals over age 80 (Cornwell et al, 1983) and have been associated with congestive heart failure, arrhythmias, and cardiac death (Hodkinson and Pomerance, 1977; Lie and Hammond, 1988). Mutant TTRs are the fibril precursor in the rare (except in some specific populations), ultimately fatal, autosomal dominant familial amyloidotic polyneuropathies (FAP) and cardiomyopathies (FAC) that have an age of onset between 30 and 60 years of age (Benson and Uemichi, 1996). In a similar fashion, the more common sporadic form of Alzheimer’s disease (AD) that appears late in life is unassociated with the mutations in AβPP or the presenilins seen in some kindreds with the rare, early-onset, autosomal dominant familial disease (Lendon et al, 1997). In contrast to atherogenesis, a process that is cumulative over the lifetime of the individual, TTR cardiac amyloid deposition and Aβ cerebral plaque formation do not appear to initiate until later in life and have been considered to reflect some aspect of the aging process. We established the transgenic model of human TTR amyloidosis to investigate the relationship between aging and in vivo amyloidogenesis, to study pathogenesis, and to develop a valid biologic system for testing potential therapies.
Results
Gene Cloning and Transgenic Strains
We cloned the normal and mutant TTR genes from the genomic DNA of an individual with a clinically aggressive form of the autosomal dominant disorder FAP (Jacobson et al, 1992). Each allele was contained on an identical 19.2 kb fragment that differed only by the diagnostic base substitution (T→C) in exon 3, accounting for the Leu→Pro amino acid substitution at position 55 (TTR Leu55Pro). The structure of the cloned genomic fragment used to produce the transgenics is shown in Figure 1. The elements known to be required for tissue-specific TTR expression are identified in the figure (Costa et al, 1988a, 1988b; Yan et al, 1990).

The human transthyretin (TTR) gene was cloned as a 19.2 kb NheI fragment, containing all the sequences known to be required for tissue specific expression, in Charon 40 grown in either Escherichia coli ED 8767 or K 802. The leu→pro substitution at amino acid 55 is encoded by a T→C transversion in the second position in the codon, located in exon 3.
Three strains were obtained with the mutant construct, two of which were lost in passage. The surviving strain carried a single copy of the mutant gene. Brother-sister matings yielded animals carrying two copies of TTR Leu55Pro. Two founders bearing the wild-type gene were identified; however, one exhibited reduced fertility and the line could not be maintained and characterized. It is not clear if this was the result of expression of the human TTR gene or related to the insertion site in the mouse genome. The surviving wild-type transgenic strain animals contained approximately 90 copies of the human insert per diploid genome (data not shown).
Human Transthyretin Transgene Expression
In both transgenic strains, the human TTR mRNA was highly expressed in the liver, eye, and brain (Fig. 2). Tissues from the animals carrying the wild-type construct contained much larger amounts of the human TTR mRNA, corresponding to the larger number of transgene copies (data not shown). With longer exposure of the Northern blots, small amounts were found in tongue, kidney, heart, skeletal muscle, and stomach. Message was not detected in other organs, including the pancreas.

Northern blot analysis of the TTR mRNA from tissues of human wild-type transthyretin transgenic mice in which 10 μg of mRNA from various tissues of transgenic mice was treated with glyoxal, separated on a 1% agarose gel, transferred onto nitrocellulose paper, and probed with 32P-cDNA of human TTR. TTR message is very abundant in liver, brain, and eye. When the autoradiographs were exposed for much longer time periods, small amounts were seen in the heart and tongue.
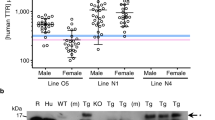
Serum TTR concentrations in the wild-type transgenics ranged between 100 and 350 mg/dl. In animals bearing the mutant construct, the amount of human TTR in the serum was between 1 and 3 mg/dl, the difference reflecting the gene dose in the two strains. (The normal human serum TTR concentration is between 10 and 40 mg/dl, [Smith and Goodman, 1971], whereas normal mouse serum contains 20 to 30 mg of murine TTR/dl of serum [Vranckx et al, 1990]). The levels increased up to 3 to 4 months of age, stabilized until 1 year, and then declined, with males showing a greater decrement than females (Table 1). The same pattern was seen when the endogenous murine serum protein was examined in the same samples.
Tissue Deposition of Human Transthyretin
None of the 157 animals examined, bearing either one (120) or two (37) copies of the mutant (TTRleu55pro) gene, developed tissue TTR deposits over observation periods as long as 2 1/2 years. In contrast, half the animals with multiple copies of the wild-type gene, between 12 and 17 months of age, showed nonfibrillar, non-Congophilic anti-human TTR-positive deposits in the kidneys, heart, or gut (Table 2). After 17 months of age, TTR-related deposits were found in 84% of the animals (89% of the males; 77% of females) bearing the wild-type human TTR gene. Although we concentrated our analyses on the cardiac and renal tissue, we also noted deposits in the lungs, tongue, and gut. No deposits were seen in the spleen or peripheral nerve. Two animals had TTR deposits in hepatic vessels.
Immunohistochemistry and Electronmicroscopy
The hearts of 16% of the animals (20% of males; 9% of females; p = .2278), mostly in animals over 2 years of age, had Congophilic deposits (Fig. 3A) that gave positive reactions with the anti-human TTR (Fig. 3B, D, F) and anti-murine SAP (Fig. 3C) antibodies and thus met the criteria for amyloid. The deposits were not reactive with anti-murine amyloid A antibody (Fig. 3E). The correspondence between the Congo red positive deposits and the immunostaining for TTR and SAP established the nature of the deposits as TTR amyloid. Figure 4, A and B shows the ultrastructural location of the myocardial deposits and the fibrillar structure typical of amyloid with an estimated fibril width of 8 to 10 nm. All 13 (10 males and 3 females) of the 83 animals older than 18 months with cardiac amyloid also had Congophilic renal deposits. One additional male had only renal amyloid.

A to F, Frozen sections of myocardium from a 22-month-old transgenic male with amyloid deposits. A, Stained with Congo red and photographed under polarization microscopy. B, An adjacent section stained with fluoresceinated anti-human TTR antibody. C and D, Adjacent sections stained with anti-murine P-component and anti-human TTR, respectively. E and F, Adjacent sections are stained with anti-murine amyloid A (E) and anti-human TTR (F). The amyloid deposits (arrows) bind Congo red and the antibodies specific for human TTR and murine serum amyloid P component (SAP). They do not react with the anti-murine amyloid A antibody, demonstrating both the nature and specificity of the staining. Original magnification: A and C to F, ×200; B, ×400.

A and B, Electron micrographs of deposits in the myocardium of the transgenic mouse shown in Figure 3. Deposits of fibrils (f), 8–10 nm in thickness and of indeterminate length, are present in the interstitium between myocyte (m) and a capillary lumen (cl). Original magnification: A, ×25,000; B, ×50,000.
In the kidney the Congophilic amyloid deposits were dense in the glomeruli and were focal in the interstitium or pelvic fat. The deposits involved all or most glomeruli (Fig. 5A), either in the mesangium or in large aggregates obliterating the glomerular tufts. The immunostains for TTR were concordant with the Congo red deposits (Fig. 5B). The amount of Congophilic material deposited in both heart and kidneys of the female mice appeared to be less than that noted in the males.

A to D, Frozen sections of kidney. A, Shows a glomerulus from the same transgenic mouse shown in 3A, stained with Congo red and photographed under polarized light. Deposits of amyloid expand the mesangium and are birefringent. B, Staining of an adjacent section with a fluoresceinated anti-human TTR antibody demonstrates deposits in capillary loops. C and D, Sections of kidney from a 22-month-old female transgenic mouse stained with Congo red (C) and anti-TTR antibody (D). There are no birefringent deposits in glomeruli stained with Congo red, but there is staining of glomerular and tubular basement membranes with the anti-TTR antibody demonstrating the immunologic identity of the deposits in male and female kidneys but the non-Congophilic nature of the deposits in the female. Original magnification: ×400.
In the majority of animals with anti-TTR-positive cardiac (14/24 males, 10/13 females) and renal (40/57 males, 27/39 females) deposits, Congo red and anti-SAP staining and electron microscopy did not reveal the characteristics of amyloid (Table 2). The non-Congophilic deposits stained for TTR in a smooth pattern along the glomerular and tubular basement membranes (Fig. 5D). In some females a few glomeruli displayed small, dense, Congophilic mesangial deposits together with the dominant non-Congophilic basement membrane staining (not shown). Some animals displayed distal tubular casts that gave positive reactions with the anti-TTR serum, indicating the presence of the human protein, an observation independent of the nature or degree of tissue deposition.
Intracellular accumulations of Congophilic or non-Congophilic aggregates were not seen in any tissue, although hepatocytes showed lightly fluorescent, diffuse, anti-TTR staining consistent with their role in TTR synthesis. One animal had Congophilic extracellular hepatic parenchymal deposits that stained with both anti-murine amyloid A (AA) and anti-human TTR antibodies. Immuno-electronmicroscopy demonstrated the anti-TTR reactivity of the fibrillar and nonfibrillar deposits with the gold-labeled second antibody (Figs. 6 and 7).

A and B, Immunoelectronmicrographs of a glomerulus from the transgenic male shown in Figure 5 A and B, after incubation with anti-TTR antibody and gold probe enhanced with silver, demonstrate gold-labeled fibrillar deposits on the luminal side of the glomerular basement membrane (bm). Original magnification: A, ×25,000; B, ×50,000.

Immuno-electron micrograph of a glomerulus from the transgenic female shown in Figure 5 C and D, after incubation with anti-TTR antibody and a 5 nm gold probe, enhanced with silver. The gold particles bind predominantly to basement membrane (arrows). Original magnification: ×50,000.
Chemical Analysis of Tissue Deposits
Extraction of Congophilic fibrils from the kidneys of a male animal using the two different methods gave the same results and revealed the fibril subunit to consist of TTR monomer with a molecular size of 14 kd, as determined by SDS-PAGE followed by Western blotting with an anti-human TTR antibody that could also react with murine TTR (Fig. 8A). Only a single band representing the human molecule was seen. Amino terminal sequence analysis of material eluted from the gel gave a single sequence GPTGTGESK, which is identical to the first nine residues of human TTR (Kanda et al, 1974). No contaminating mouse TTR sequence was seen. The isolated protein was subjected to mass spectroscopy with the results confirming that the protein was unmodified human TTR monomer. Although less protein could be extracted from the non-Congophilic deposits, the results on Western blotting (Fig. 8A), mass spectroscopy, and amino-terminal sequence analysis were identical to those obtained with the fibrillar deposits, indicating that the primary structure of the nonfibrillar protein also consisted of intact human TTR. Kidney extracts from an animal with a high level of human TTR in its serum, but no tissue deposits, yielded no detectable TTR immunoreactive material, indicating that the material extracted from the positive tissues was not the result of an absorption artifact (Fig. 8A, lane 5).

A, Western blotting of proteins extracted from kidneys of mice transgenic for the human TTR gene using the acetonitrile/trifluoracetic acid extraction procedure (Kaplan et al, 1993). The kidneys were those shown in Figure 5. Lane 1 shows material from a female mouse with non-Congophilic deposits. Lane 2 contains an extract from a transgenic female mouse with Congo red positive deposits. Lane 3 has the same preparation from a transgenic male mouse with non-Congophilic deposits. Lane 4 displays material from a transgenic male in which the deposits were Congo red-positive. Extraction of material from renal tissue of a transgenic female mouse with a serum human TTR concentration of 170 mg/dl, but no immunohistochemically detectable tissue deposition, yielded no immunoreactive material (lane 5). Markers (21 and 14 kd) are shown in the extreme left hand lane. B, An extract prepared from the heart of an animal with Congo red-positive deposits is shown in lane 2. Lane 1 contains a recombinant human TTR control, whereas lane 3 shows an extract of liver from a transgenic animal without deposits, representing material being synthesized by hepatocytes at the time of sacrifice. All three preparations show anti-human TTR reactive bands of the size of the intact monomer. Lane 4 contained a sample of bovine serum albumin as a protein control.
Western blot analysis of material extracted from an amyloid-containing heart also showed a single immunoreactive species, that comigrated with both intact recombinant TTR and soluble TTR synthesized in the liver (Fig. 8B). There was no evidence for a smaller TTR-related peptide nor the more rapidly migrating mouse TTR.
Discussion
To study amyloidogenesis and examine factors that enhance or inhibit fibril formation by amyloidogenic substrates in vivo, we produced several strains of mice transgenic for different forms of the human TTR gene. We were able to maintain and study two of these strains, one bearing a single integrated copy of a gene encoding a highly amyloidogenic variant protein and the other carrying many copies of the wild-type allele. Animals carrying one or two copies of the human TTR pro55 allele did not develop detectable TTR-related deposits in the course of 2 1/2 years of observation. During the same period, a significant number of animals in the strain producing large amounts of the normal sequence human protein displayed cardiac amyloid deposition that resembles that seen in human senile cardiac amyloidosis with respect to both tissue specificity and its occurrence late in life. However, the most common pathologic finding in animals bearing the wild-type gene was the nonfibrillar and non-Congophilic deposition of intact human TTR molecules in the myocardium and renal parenchyma.
The data demonstrate that mice transgenic for genes encoding wild-type human TTR provide a valid model of human senile systemic (cardiac) amyloidosis. The myocardial deposits are multifocal in the ventricles, age-dependent, occurring in late adult life, and, in some experiments, more common in males than females. Whereas wild-type TTR renal amyloid has not been noted in humans, deposition of TTR amyloid in the kidneys has been observed in some Portuguese kindreds bearing the TTR val30 met mutation (Lobato et al, 1998).
The nonamyloid deposits have no TTR counterpart in clinical material. In elderly humans, the detection of nonamyloid TTR deposits requires immunohistochemical staining of tissues with a specific antibody. Because this is not done, the identification of such deposits in the course of routine histologic examination is unlikely, rendering the prevalence of potentially pathogenic non-Congophilic TTR deposits in elderly humans unknown. The deposits in the mice may be analogous to the nonfibrillar deposits seen in human monoclonal immunoglobulin (Ig) light-chain deposition disease, the diffuse Aβ plaques seen in patients with AD, or findings reported in a dialysis patient with systemic, nonamyloidotic β2 microglobulin deposition (Bugiani et al, 1989; Buxbaum et al, 1990; Terreros et al, 1989). In the Ig light-chain instance, most patients have either Congophilic or non-Congophilic deposits, leading to the speculation that the nature of the deposits is a function of the primary structure of protein precursor. A few patients have been described in which both types of deposits have been noted. In one such patient, identity of size and amino-terminal sequence has been demonstrated in the L-chain molecules extracted from both types of deposits (Kaplan et al, 1997). In Aβ deposition the diffuse and hard plaques were noted concurrently, and the assumption was made that the diffuse deposits are a precursor.
In studies unrelated to amyloid deposition, both rat and human proximal renal tubule cells have given positive immunologic reactions with antibodies specific for TTR. It has been hypothesized that the staining represents molecules reabsorbed or undergoing degradation by the tubular epithelium (Kato et al, 1984; Makover et al, 1988; Tsuzuki et al, 1997). It is possible that the tubular staining might be accentuated in animals where a large TTR load is presented to the kidneys. Our data do not support such a notion because the staining pattern in those studies differed from the basement membrane localization seen in the TTR transgenics.
Earlier work has shown that transgenic mice bearing 30 to 60 copies of the mutant TTR met30 gene developed Congophilic deposits in the heart and kidneys (Kohno et al, 1997). In that strain, amyloid was first detected in the gut beginning at 5 to 6 months of age, far earlier than in our experiments. The Congo red staining material was also reactive with antibodies specific for human TTR and murine SAP. The investigators did not report the presence of non-Congophilic deposition in any organ. The failure to identify nonfibrillar TTR deposits may have been technical or may reflect differences in the particular transgene, its site of genomic integration, differences in background genes present in the recipient strains, or other unknown factors. When the multicopy TTR locus was bred onto a TTR knockout background, no difference was seen in the frequency or pace of amyloid deposition, indicating that the mouse protein had no effect.
The non-Congophilic deposits in our animals may represent either a preamyloid intermediate state or the product of an alternative path of processing of the same substrate. The appearance of only non-Congophilic deposits between 12 and 18 months of age suggests, but does not prove, a precursor role. Similarly, the coexistence of small Congo red positive deposits in the kidneys of some animals with preponderantly nonfibrillar deposition may indicate a precursor role, perhaps dependent on the tissue site or concentration of substrate.
In vitro assays of fibrillogenesis with soluble precursors of both mutant and normal sequence forms of TTR have suggested that the fibril precursor is primarily a TTR monomer that assumes an amyloidogenic configuration during unfolding at low pH, a phenomenon more likely to occur during the catabolism of the protein in a lysosomal cell compartment than during its synthesis (Colon and Kelly, 1992). Other investigators have reported that a misfolded monomeric species can be detected at neutral pH and presented evidence for aggregate formation from the isolated monomers. The latter observation is more consistent with the extracellular localization of TTR deposits in vivo; however, actual fibril formation was not demonstrated (Quintas et al, 1999). The data from our transgenic animals are consistent with the in vitro observations but suggest that the steps of deposition and fibril formation may be dissociated and thus require more detailed analysis, particularly with respect to intermediate forms of aggregation. In these animals neither process appears to require proteolysis of the native TTR.
The in vitro experiments cannot account for the temporal delay in the deposition of precursor on the basis of an age-related change in the primary structure of TTR. Both forms of deposition seem to be identical with respect to primary sequence. It has been suggested that serum TTR is modified by sulfation with increasing age, and this modification could account for its propensity to deposit (Kishikawa et al, 1999; Suhr et al, 1999). Further studies are in progress in the transgenic animals to formally evaluate the possibility.
Our observations, as well as the occurrence of non-Congophilic Aβ diffuse plaques and prion protein (PrP) deposits, support the notion that, in vivo, the process could be stepwise, with other factors mediating the formation of the highly ordered, more biologically stable Congophilic fibril. Comparable findings were noted when an Aβ transgene was bred into a strain lacking genes for apolipoprotein E (ApoE); virtually no thioflavine S-positive plaques were seen in the hippocampus. Instead, the animals showed only sparse immunoreactive Aβ deposits with the characteristics of diffuse plaques, an observation consistent with the findings in the younger TTR transgenics (Bales et al, 1997).
Transgenic mice with other forms of amyloid deposition have been reported (Hoppener et al, 1993; Prusiner, 1991; Seabrook and Rosahl, 1999). In all of the models, it seems that over-expression of the precursor, whether it is wild-type or mutant sequence, is critical. The in vivo data suggest that any hypothesis regarding fibrillogenesis generated by the in vitro studies should be placed in the context of a more biologic “equilibrium hypothesis” encompassing factors influencing the synthetic and catabolic rates of the precursor, which are likely to determine the number of molecules available to populate the protofibril pool and its interaction with other small and large molecules, perhaps ApoE, SAP, or heparan sulfate proteoglycan. Our data confirm that SAP is only found in fibrillar deposits. They also suggest that deposition precedes SAP binding, because TTR deposition is seen at a younger age at the same sites subsequently found to display fibril deposition and SAP binding. The observations in these TTR transgenic mice clearly indicate that a variety of interactions between amyloid precursors and other cellular and molecular elements in vivo impact on the events demonstrable in test tube fibrillogenesis (Teng and Buxbaum, 1996). The model will allow us to define those interactions and determine whether the phenomena are particular to TTR or occur, to some extent, in all forms of amyloidosis.
Materials and Methods
Wild-type and mutant human TTR genes were cloned from genomic DNA extracted from lymphoid cells immortalized from a patient with FAP secondary to a substitution of proline for leucine at position 55 (Jacobson et al, 1992). NheI fragments (19.2 kb) were inserted into Charon 40 and grown on either Escherichia coli ED 8767 or K802 (Sambrook et al, 1989). The insert contained the entire gene, with all the elements known to be required for tissue-specific expression. It was isolated from the vector by Not I, Sca I digestion and injected into fertilized ova from C57Bl/6 x DBA/2 (B6D2) F2 animals. The transfected ova were placed in the uteri of pseudopregnant B6D2 F1 females (Hogan et al, 1986). DNA was extracted from tail tissue of the offspring and amplified using human TTR exon-specific primers to determine if the complete human gene was present (Jacobson et al, 1988). The amplification products were analyzed by agarose gel electrophoresis. Gene copy number was determined by slot blotting using human genomic DNA as a standard (Warren et al, 1987). Tissue expression was assayed by Northern blotting of RNA extracted from individual tissues (Teng et al, 1989). Serum levels of human TTR were measured by an ELISA using an anti-human TTR antibody with dilutions of purified recombinant human TTR (provided by Dr. Jeffrey Kelly, The Scripps Research Institute, La Jolla, California) used to construct a standard curve (Matsumoto et al, 1993).
Animals were fed standard rodent chow with water provided ad libitum. They were maintained in an Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC)-accredited specific-pathogen-free (SPF) animal facility with no more than five animals in a cage.
Tissues were prepared for histologic examination from animals killed by pentobarbital euthanasia or, rarely, from animals found dead in cages. Sections of snap-frozen tissues (heart, liver, kidneys from all animals; spleen, stomach, small and large intestine, abdominal fat, tongue, lung, skeletal muscle, and peripheral nerve from some animals) were incubated separately with the following unlabeled antibodies: goat IgG fraction of anti-human TTR (Atlantic Antibodies, Stillwater, Minnesota) and/or rabbit anti-human TTR (Dako, Carpinteria, California), rabbit anti-mouse amyloid A (gift of Dr. Jean Sipe, Department of Biochemistry, Boston University School of Medicine, Boston, Massachusetts), and rabbit anti-mouse SAP (Calbiochem, San Diego, California), followed by fluorescein-conjugated rabbit anti-goat immunoglobulin or swine anti-rabbit immunoglobulin (Dako), according to established methods in our laboratory (Gallo et al, 1986).
Polarization microscopy of Congo red stained sections was performed on the same section used for immunofluorescence or an adjacent section of the same tissue specimen. Tissues were examined in parallel with those of age- and sex-matched nontransgenic littermates with the origin of the tissues unknown to the examiner. As expected, some older nontransgenic animals and a similar number of their transgenic littermates had Congophilic tissue deposits that stained for AA amyloid, reflecting the known incidence of this phenomenon in various mouse strains (Table 2) (Lipman et al, 1993). The distribution and amount of TTR amyloid deposition were noted and graded semiquantitatively.
For electron microscopy, 1- to 3-mm blocks of frozen tissue from selected animals were fixed in cold 4% paraformaldehyde and 0.2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) overnight at 4° C. After fixation, the pieces were washed three times in buffer and progressively dehydrated in ethanol (50%, 70%, 95%, and 100% for 30 minutes per step). They were then incubated in a 2:1, then 1:2, mixture of ethanol and Unicryl (Vector Laboratories, Burlingame, California), for 30 minutes each, followed by another 60 minutes, and then an overnight infiltration in pure resin, all at −20° C. This was followed by polymerization at 40 to 45° C for 3 to 4 days.
For immuno-electronmicroscopy, ultrathin sections of Unicryl-embedded tissue mounted on nickel grids were preincubated for 30 minutes on drops of normal goat serum (Dako, Glostrup, Denmark), followed by an overnight incubation with rabbit anti-human TTR or control rabbit serum diluted 1:10, in a humidified chamber at 4° C. The grids were extensively washed with tris-buffered saline (TBS), then incubated for 2 hours at room temperature with 5 or 10 nm gold-labeled anti-rabbit IgG (Dako), and diluted 1:20. They were then washed in TBS and double-distilled water, briefly stained with uranyl acetate and lead citrate, and examined with a Zeiss EM-10 electron microscope. In some experiments improved visualization of the 5 nm gold particles was obtained by silver enhancement (BB International, Cardiff, United Kingdom).
Protein extraction and purification were carried out on approximately 100 mg of kidney (from transgenic animals with amyloid and nonamyloid TTR deposits or no deposits and nontransgenic littermate controls), cut into1- to 3-mm pieces in iced saline in a cocktail of protease inhibitors (PI) (1μm pepstatin, 100 μm TLCK-HCl, and 1 μm Leupeptin, all from Boehringer-Mannheim [now Roche Diagnostics] Indianapolis, Indiana) and homogenized on ice. After centrifugation at 12,000 rpm for 15 minutes at 4° C, the material was washed four times in the presence of PI and three times in saline without PI. After centrifugation as above, the resulting pellet was resuspended in 20 volumes of 2 mm CaCl2 in 0.1 M Tris-HCl (pH 7.5) and 3 mm NaN3, and digested with a mixture of 2 mg/ml collagenase CLS-3 (Worthington Biochemicals, Lakewood, New Jersey) and 10 μg/ml DNase I (Worthington Biochemicals) for 18 hours at 37° C. After digestion, the suspension was centrifuged at 12,000 rpm for 10 minutes at 4° C, washed three times with saline containing PI and incubated for 1 hour at room temperature with either 99% formic acid (Sigma, St. Louis, Missouri) or with 20% acetonitrile/0.1% trifluoracetic acid. After centrifugation at 12,000 rpm for 5 minutes, the supernatant was dried under N2 and analyzed by Tris-Tricine SDS-PAGE on 16% gels and Western blotting (Kaplan et al, 1993; Roher et al, 1993).
After the extracted proteins were electrophoresed, they were transferred electrophoretically to polyvinylidene difluoride membranes (PVDF, Immobilon; Millipore Corporation, Bedford, Massachusetts) for 1 hour at 400 mA at 4° C using 10 mm 3 cyclohexylamino-1-propanesulfonic acid (CAPS) buffer, pH 11, containing 10% methanol. The membranes were blocked by incubating with 5% nonfat milk in 10 mm phosphate buffer, 137 mm NaCl, 2.7 mm Kcl, pH 7.4 (PBS), containing 0.1% Tween-20 (PBS-T). They were incubated for 2 hours at room temperature with the first antibody (either rabbit anti-human TTR (Dako) diluted 1:1,000 in PBS-T or rabbit anti-human TTR (Boehringer-Mannheim) diluted 1:4,000 in PBS-T. The Dako antibody was also reactive with murine TTR on Western blots. As a second antibody, we used horseradish peroxidase-linked F(ab’)2 fragment of donkey anti-rabbit Ig 1:5,000 in PBS-T. Immunoblots were visualized by chemiluminescence according to the manufacturer’s instructions (Amersham Pharmacia Biotech, Piscataway, New Jersey).
For sequencing, proteins electrotransferred onto PVDF membranes were stained with 0.5% Coomassie blue R-250 (BioRad, Hercules, California), and the bands were excised. Sequencing was carried out using automated Edman degradation on a 477A protein-peptide sequencer. The resulting phenylthiohydantoin amino acid derivatives were identified using the online 120A PTH analyzer (Applied Biosystems, Foster City, California).
For mass spectroscopy the acetonitrile/trifluoracetic acid extracts were resuspended in PBS and incubated overnight at 4° C with 50 μl of paramagnetic beads M-450 anti-rabbit IgG (Dynal, Oslo, Norway) previously coated with 3 μl polyclonal rabbit anti-human TTR antibodies (Dako). After the incubation, the beads were washed three times with PBS and eluted with 10 μl isopropyl alcohol, formic acid mixture (4:4:1) and dried completely in a Speed-vac concentrator. Linear time-of-flight mass spectroscopy was carried out at the W. M. Keck facility at Yale University, New Haven, Connecticut.
References
Bales KR, Verina T, Dodel RC, Du Y, Altstiel L, Bender M, Hyslop P, Johnstone EM, Little SP, Cummins DJ, Piccardo P, Ghetti B, and Paul SM (1997). Lack of apolipoprotein E dramatically reduces amyloid beta-peptide deposition. Nat Genet 17: 263–264.
Benson MD and Uemichi T (1996). Transthyretin amyloidosis. Amyloid Int J Exp Clin Invest 3: 44–56.
Bugiani O, Giaccone G, Frangione B, Ghetti B, and Tagliavini F (1989). Alzheimer patients: Preamyloid deposits are more widely distributed than senile plaques throughout the central nervous system. Neurosci Lett 103: 263–268.
Buxbaum JN, Chuba JV, Hellman GC, Solomon A, and Gallo GR (1990). Monoclonal immunoglobulin deposition disease: Light chain and light and heavy chain deposition diseases and their relation to light chain amyloidosis. Clinical features, immunopathology, and molecular analysis. Ann Intern Med 112: 455–464.
Buxbaum JN and Tagoe CE (2000). The genetics of the amyloidoses. Annu Rev Med 51: 543–569.
Colon W and Kelly JW (1992). Partial denaturation of transthyretin is sufficient for amyloid fibril formation in vitro. Biochemistry 31: 8654–8660.
Cornwell GG III, Murdoch WL, Kyle RA, Westermark P, and Pitkanen P (1983). Frequency and distribution of senile cardiovascular amyloid. A clinicopathologic correlation. Am J Med 75: 618–623.
Costa RH, Grayson DR, Xanthopoulos KG, and Darnell JE Jr (1988a). A liver-specific DNA-binding protein recognizes multiple nucleotide sites in regulatory regions of transthyretin, alpha 1-antitrypsin, albumin, and simian virus 40 genes. Proc Natl Acad Sci USA 85: 3840–3844.
Costa RH, Lai E, Grayson DR, and Darnell JE Jr (1988b). The cell-specific enhancer of the mouse transthyretin (prealbumin) gene binds a common factor at one site and a liver-specific factor(s) at two other sites. Mol Cell Biol 8: 81–90.
Gallo GR, Feiner HD, Chuba JV, Beneck D, Marion P, and Cohen DH (1986). Characterization of tissue amyloid by immunofluorescence microscopy. Clin Immunol Immunopathol 39: 479–490.
Hodkinson HM and Pomerance A (1977). The clinical significance of senile cardiac amyloidosis: A prospective clinico-pathological study. Q J Med 46: 381–387.
Hogan B, Constantini F, and Lacy E (1986). Manipulating the mouse embryo: A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory, 89–107, 157–173.
Hoppener JW, Verbeek JS, de Koning EJ, Oosterwijk C, van Hulst KL, Visser-Vernooy HJ, Hofhuis FM, van Gaalen S, Berends MJ, and Hackeng WH (1993). Chronic overproduction of islet amyloid polypeptide/amylin in transgenic mice: lysosomal localization of human islet amyloid polypeptide and lack of marked hyperglycemia or hyperinsulinaemia. Diabetologia 36: 1258–1265.
Jacobson DR, McFarlin DE, Kane I, and Buxbaum JN (1992). Transthyretin Pro55, a variant associated with early-onset, aggressive, diffuse amyloidosis with cardiac and neurologic involvement. Hum Genet 89: 353–356.
Jacobson DR, Pras M, McFarlin DE, and Buxbaum J (1988). Two new DNA-based tests for mutations causing familial amyloidotic polyneuropathy. In: Isobe T, Araki S, Uchino F, Kito S, and Tsubura E, editors. Amyloid and amyloidosis. New York: Plenum, 301–305.
Kanda Y, Goodman DS, Canfield RE, and Morgan FJ (1974). The amino acid sequence of human plasma prealbumin. J Biol Chem 249: 6796–6805.
Kaplan B, German G, and Pras M (1993). Isolation and characterization of amyloid proteins using milligram amounts of amyloid-containing tissue. J Liquid Chromatogr 11: 2249–2268.
Kaplan B, Vidal R, Kumar A, Ghiso J, Frangione B, and Gallo G (1997). Amino-terminal identity of co-existent amyloid and non-amyloid immunoglobulin kappa light chain deposits. A human disease to study alterations of protein conformation. Clin Exp Immunol 110: 472–478.
Kato M, Kato K, and Goodman DS (1984). Immunocytochemical studies on the localization of plasma and of cellular retinol-binding proteins and of transthyretin (prealbumin) in rat liver and kidney. J Cell Biol 98: 1696–1704.
Kelly JW and Lansbury PT Jr (1994). A chemical approach to elucidate the mechanism of transthyretin and beta-protein amyloid fibril formation. Amyloid Int J Exp Clin Invest 1: 186–205.
Kishikawa M, Nakanishi T, Miyazaki A, and Shimizu A (1999). Enhanced amyloidogenicity of sulfonated transthyretin in vitro, a hypothetical etiology of senile amyloidosis. Amyloid 6: 183–186.
Kohno K, Palha JA, Miyakawa K, Saraiva MJ, Ito S, Mabuchi T, Blaner WS, Iijima H, Tsukahara S, Episkopou V, Gottesman ME, Shimada K, Takahashi K, Yamamura K, and Maeda S (1997). Analysis of amyloid deposition in a transgenic mouse model of homozygous familial amyloidotic polyneuropathy. Am J Pathol 150: 1497–1508.
Lendon CL, Ashall F, and Goate AM (1997). Exploring the etiology of Alzheimer disease using molecular genetics. JAMA 277: 825–831.
Lie JT and Hammond PI (1988). Pathology of the senescent heart: Anatomic observations on 237 autopsy studies of patients 90 to 105 years old. Mayo Clin Proc 63: 552–564.
Lipman RD, Gaillard ET, Harrison DE, and Bronson RT (1993). Husbandry factors and the prevalence of age-related amyloidosis in mice. Lab Anim Sci 43: 439–444.
Lobato L, Beirao I, Guimaraes SM, Droz D, Guimaraes S, Grunfeld JP, and Noel LH (1998). Familial amyloid polyneuropathy type I (Portuguese): Distribution and characterization of renal amyloid deposits. Am J Kidney Dis 31: 940–946.
Makover A, Moriwaki H, Ramakrishnan R, Saraiva MJM, Blaner WS, and Goodman DS (1988). Plasma Transthyretin. Tissue sites of degradation and turnover in the rat. J Biol Chem 263: 8598–8603.
Matsumoto T, Morita M, Shirai H, and Kishi T (1993). Construction of sandwich enzyme immunoassay for rat transthyretin using recombinant antigen approach, and its application. Biosci Biotechnol Biochem 57: 414–418.
Prusiner SB (1991). Molecular biology and transgenetics of prion diseases. Crit Rev Biochem Mol Biol 26: 397–438.
Quintas A, Saraiva MJ, and Brito RM (1999). The tetrameric protein transthyretin dissociates to a non-native monomer in solution. A novel model for amyloidogenesis. J Biol Chem 274: 32943–32949.
Roher AE, Lowenson JD, Clarke S, Woods AS, Cotter RJ, Gowing E, and Ball MJ (1993). beta-Amyloid-(1–42) is a major component of cerebrovascular amyloid deposits: implications for the pathology of Alzheimer disease. Proc Natl Acad Sci USA 90: 10836–10840.
Sambrook J, Fritsch EF, and Maniatis T (1989). Molecular Cloning: A Laboratory Manual, 2nd ed. Cold Spring Harbor Laboratory, Cold Spring Harbor, NY: 2.11–2.125.
Seabrook GR and Rosahl TW (1999). Transgenic animals relevant to Alzheimer’s disease. Neuropharmacology 38: 1–17.
Smith FR and Goodman DS (1971). The effects of diseases of the liver, thyroid, and kidneys on the transport of vitamin A in human plasma. J Clin Invest 50: 2426–2436.
Suhr OB, Svendsen IH, Ohlsson PI, Lendoire J, Trigo P, Tashima K, Ranlov PJ, and Ando Y (1999). Impact of age and amyloidosis on thiol conjugation of transthyretin in hereditary transthyretin amyloidosis. Amyloid 6: 187–191.
Teng MH and Buxbaum JN (1996). Transgenic mice in amyloid research. Amyloid 3: 187–208.
Teng MH, Hedayati S, Alexander AA, Barkin R, Basch RS, and Buxbaum JN (1989). A Thy-1 negative lymphoma cell variant defective in the formation of glycosyl-phosphatidylinositol membrane protein anchors. Mol Immunol 26: 391–402.
Terreros DA, Knight JA, Peric-Golia L, and Cheung AK (1989). Generalized β2-microglobulin deposition. A preamyloidosis disorder? Archives of Pathology & Laboratory Medicine 113: 31–35.
Tsuzuki K, Fukatsu R, Hayashi Y, Yoshida T, Sasaki N, Takamaru Y, Yamaguchi H, Tateno M, Fujii N, and Takahata N (1997). Amyloid beta protein and transthyretin, sequestrating protein colocalize in normal human kidney. Neurosci Lett 222: 163–166.
Vranckx R, Savu L, Maya M, Rouaze-Romet M, and Nunez EA (1990). Immunological quantitation of rat and mouse thyroxine-binding globulins. Ontogenesis and sex-dependence of the circulating levels of the thyroxine-binding globulins. Acta Endocrinol 123: 649–656.
Warren AC, Robakis NK, Ramakrishna N, Koo EH, Ross CA, Robb AS, Folstein MF, Price DL, and Antonarakis SE (1987). beta-Amyloid gene is not present in three copies in autopsy-validated Alzheimer’s disease. Genomics 1: 307–312.
Wright JR and Calkins E (1981). Clinical-pathologic differentiation of common amyloid syndromes. Medicine 60: 429–448.
Yan C, Costa RH, Darnell JE Jr, Chen JD, and Van Dyke TA (1990). Distinct positive and negative elements control the limited hepatocyte and choroid plexus expression of transthyretin in transgenic mice. EMBO J 9: 869–878.
Acknowledgements
The authors acknowledge the assistance of Dr. Elizabeth Newcomb of the New York University School of Medicine Transgenic Facility in the production of the transgenic strains. The authors also gratefully acknowledge the assistance of Dr. Herman Yee in the Department of Pathology at Bellevue Hospital and the outstanding efforts of Ms. Kathy Maxwell at the Scripps Research Institute in the preparation of the material for publication.
The studies were partially supported by NIH grants RO1DK34900, RO1AG15916, and RO1AG05891 (RV), Merit Review Funds from the Department of Veterans Affairs, and the New York University School of Medicine Honors Program (AS, RR).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Teng, Mh., Yin, Jy., Vidal, R. et al. Amyloid and Nonfibrillar Deposits in Mice Transgenic for Wild-Type Human Transthyretin: A Possible Model for Senile Systemic Amyloidosis. Lab Invest 81, 385–396 (2001). https://doi.org/10.1038/labinvest.3780246
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1038/labinvest.3780246
This article is cited by
-
Amyloid deposition in a mouse model humanized at the transthyretin and retinol-binding protein 4 loci
Laboratory Investigation (2018)
-
Relevance of mouse models of cardiac fibrosis and hypertrophy in cardiac research
Molecular and Cellular Biochemistry (2017)
-
Human TTRV30M localization within podocytes in a transgenic mouse model of transthyretin related amyloidosis: does the environment play a role?
Transgenic Research (2013)
-
Aged vervet monkeys developing transthyretin amyloidosis with the human disease-causing Ile122 allele: a valid pathological model of the human disease
Laboratory Investigation (2012)
-
Transthyretin: the servant of many masters
Cellular and Molecular Life Sciences (2009)
