
Abstract
Historic Japanese culture evolved from at least two distinct migrations that originated on the Asian continent. Hunter-gatherers arrived before land bridges were submerged after the last glacial maximum (>12,000 years ago) and gave rise to the Jomon culture, and the Yayoi migration brought wet rice agriculture from Korea beginning ~2,300 years ago. A set of 81 Y chromosome single nucleotide polymorphisms (SNPs) was used to trace the origins of Paleolithic and Neolithic components of the Japanese paternal gene pool, and to determine the relative contribution of Jomon and Yayoi Y chromosome lineages to modern Japanese. Our global sample consisted of >2,500 males from 39 Asian populations, including six populations sampled from across the Japanese archipelago. Japanese populations were characterized by the presence of two major (D and O) and two minor (C and N) clades of Y chromosomes, each with several sub-lineages. Haplogroup D chromosomes were present at 34.7% and were distributed in a U-shaped pattern with the highest frequency in the northern Ainu and southern Ryukyuans. In contrast, haplogroup O lineages (51.8%) were distributed in an inverted U-shaped pattern with a maximum frequency on Kyushu. Coalescent analyses of Y chromosome short tandem repeat diversity indicated that haplogroups D and C began their expansions in Japan ~20,000 and ~12,000 years ago, respectively, while haplogroup O-47z began its expansion only ~4,000 years ago. We infer that these patterns result from separate and distinct genetic contributions from both the Jomon and the Yayoi cultures to modern Japanese, with varying levels of admixture between these two populations across the archipelago. The results also support the hypothesis of a Central Asian origin of Jomonese ancestors, and a Southeast Asian origin of the ancestors of the Yayoi, contra previous models based on morphological and genetic evidence.
Similar content being viewed by others

Introduction
After more than 100 years of intense investigation (Mizoguchi 1986), the origin of the Japanese population still remains controversial. A number of contrasting models attempt to explain the fairly large range of morphological, cultural, and genetic variation represented in modern Japanese populations and recognized in the archaeological and fossil records. Abundant archaeological evidence documents at least two major migration processes that brought people from the Asian continent to the Japanese archipelago. An early wave of migration brought people to Japan more than 30,000 years ago (Ono et al. 2002; Takamiya 2002), and gave rise to the Jomon culture ≥12,000 years ago (Chard 1974). The Jomon people lived a hunter-gatherer lifestyle and made some of the world’s earliest pottery for several thousand years in isolation before a second major wave of migration began. Entering from the Korean Peninsula around 2,300 years ago, the Yayoi migration brought wet rice agriculture, weaving, and metalworking to Japan (Chard 1974). The effects of this new culture are first seen in Kyushu, and then spread northeastward. By about 300 a.d. Yayoi culture completed its expansion, resulting in the alteration of all local cultures south of Hokkaido. It is generally accepted that modern Japanese are descendants of the two populations who produced the Jomon and the Yayoi cultures. However, the relative genetic contribution each of these populations made to mainland Japanese, and the geographic regions from where they originated on continental Asia are still in dispute.
There are two contemporary ethnic groups that are distinct from Japanese populations living on the centrally located islands of Honshu, Shikoku and Kyushu (Fig. 1). These are the Ainu, inhabiting the northern island of Hokkaido, and the Ryukyuans living on the southernmost islands, including Okinawa. The Ryukyuans and Ainu are generally recognized as remnant populations descended from the Jomon, with the Ainu surviving in Hokkaido in relative isolation from post-Jomon influences until the end of the last century (Howells 1966; Omoto and Misawa 1976; Turner 1976; Ossenberg 1986; Hanihara 1991). Like many debates on human evolution, current hypotheses on the origins of the mainland Japanese can be classified into three groups: replacement, hybridization, and transformation (Suzuki 1981). The first model involves complete replacement of Jomon genetic lineages by those of the Yayoi (Howells 1966; Turner 1976). Transformation models contend that genetic variation in modern Japanese populations derives solely from Jomon ancestors and does not reflect Yayoi admixture (Suzuki 1981; Mizoguchi 1986). The hybridization model claims that modern Japanese are the result of admixture between the different immigrant populations, with contemporary variation reflecting different degrees of admixture between Jomon and Yayoi people. Both the replacement (Cavalli-Sforza et al. 1994) and the transformation (Nei 1995) models have been supported by genetic data, with more recent support emerging in favor of the hybridization model (Hammer and Horai 1995; Horai et al. 1996; Omoto and Saitou 1997; Sokal and Thomson 1998; Tanaka et al. 2004).

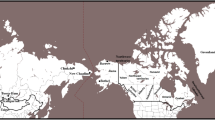
Map showing the approximate geographic positions of 39 populations sampled in this study. The populations are grouped into four major geographic areas (dashed circles surround Central, Southeastern, and Northeastern Asians). Three letter codes are as follows: Japan: AIN Ainu, AOM Aomori, SHI Shizuoka, TOK Tokushima, KYU Kyushu, OKI Okinawa; Northeast Asia: KOR Korea, NHA northern Han, MAN Manchu, MEV Manchurian Evenk, BUR Buryat, EVK Evenk, EVN Even, ORO Oroqen; Southeast Asia: TAI Taiwan Han and Hakka, TAB Taiwan Aboriginal, SHA Southern Han, TUJ Tujia, YIZ Yizu, MIA Miao, YAO Yao, ZHU Zhuang, SHE She, VIE Vietnam, MAL Malay, FIL Philippines, INW Indonesia West; Central Asia: UYG Uygur, MON Mongolia, ALT Altai, TIB Tibet; South Asia: IND India and SRI Sri Lanka; Oceania: INE Indonesia East, AUS Australian aboriginal people, PNG Papua New Guinea, MEL Melanesia, MIC Micronesia, POL Polynesia. The sampling locations of four populations (EVN, MIC, POL, MEL) are not on the map
Controversy also surrounds the origins of Japanese lineages before they arrived on the archipelago. While the Yayoi are generally believed to have come from northeast Asia by way of Korea (Chard 1974; Sokal and Thomson 1998), there is a longstanding controversy on the origin of the Jomon. Inferences based on dental (Turner 1976, 1990) and cranial morphology (Hanihara 1991) have been used to suggest that the Jomon people are direct descendants of Southeast Asians who migrated to Japan when it was still connected to the Asian mainland. There were Pleistocene land bridges connecting Japan to the mainland both in the north by way of Hokkaido and Sakhalin Island, and in the south by way of Korea (Aikens and Higuchi 1982). On the other hand, several authors have claimed support for the hypothesis of a northeast Asian origin of the Jomon, with classical marker polymorphisms revealing Japanese affinities with north Asian populations (Nei 1995; Bannai et al. 1996; Matsumoto et al. 1997; Omoto and Saitou 1997), and Y chromosomal and mitochondrial DNA (mtDNA) studies exhibiting closer connections of Japanese with Koreans and Tibetans (Hammer and Horai 1995; Horai et al. 1996; Tajima et al. 2002, 2004).
The male-specific portion of the Y chromosome has proven to be an extremely useful tool for tracing paternal ancestry. As a result of its lack of recombination and multiple marker systems, this haploid system provides the opportunity to infer demographic processes that have occurred over a range of time scales (Jobling and Tyler-Smith 2003). Previously, Hammer and Horai (1995) surveyed a small number of Y-linked polymorphisms and hypothesized that both the Jomon and Yayoi cultures made significant genetic contributions to extant Japanese populations. Here, we further address this hypothesis, and investigate the origins of Jomon and Yayoi male lineages before they arrived in Japan, through a large-scale survey of Japanese and Asian populations using a battery of 81 Y-linked binary polymorphisms. We also analyze a set of ten Y-short tandem repeats (STRs) in the same samples to provide a temporal framework for the origin of Japanese founder Y chromosomes.
Materials and methods
Subjects and Y chromosome terminology
We examined 259 individuals from six Japanese populations representing key geographic areas: Okinawa, Kyushu, Tokushima, Shizuoka, Aomori, and Hokkaido (Fig. 1). In addition, we included a total of 2,248 males from 33 populations. These populations are subdivided into regional groups: Northeast Asia, Southeast Asia, Central Asia, and South Asia, and Oceania (Fig. 1). Many samples analyzed here were included in previous studies (Hammer and Horai 1995; Hammer et al. 2001; Karafet et al. 2001, 2005; Redd et al. 2002b). All sampling protocols were approved by the Human Subjects Committee at the University of Arizona. We follow the terminological conventions recommended by the Y Chromosome Consortium (YCC 2002) for naming NRY lineages. Capital letters A–R are used to identify the 18 major Y chromosome clades or haplogroups. Lineages not defined on the basis of a derived character state represent interior nodes of the tree (shown as dotted lines in Fig. 2) are potentially paraphyletic (Hammer and Zegura 2002). These “paragroups” are distinguished from haplogroups by the asterisk symbol. For convenience we refer to paragroups as haplogroups or lineages throughout the text. When no farther downstream markers in the latest YCC tree (Jobling and Tyler-Smith 2003) are typed, we designate haplogroups by a capital letter corresponding to the Y chromosome clade and the name of the most derived typed marker. Distinct Y chromosomes identified by STRs are designated as “haplotypes” (de Knijff 2000).
Genetic markers
The polymorphic sites in the present study included a set of 71 previously published binary Y chromosome markers (Karafet et al. 2005). Additionally, we genotyped seven polymorphisms (M25, M73, M55, M116, M124, M269, and M178) (Underhill et al. 2000; Cruciani et al. 2002). We also typed three novel polymorphisms within the arylsulfatase D-pseudogene (ARSDP) that were discovered in a panel of 92 Y chromosomes from sub-Saharan Africa, Asia, Europe, Oceania, and the Americas (Hammer et al. 2003). These mutations (referred to as P42, P47, and P49, respectively) mark sub-clades of common Japanese haplogroups: a G→A transition at position 57,713, a C→T transition at position 73,152 and an A→T transversion at position 72,392. Primer sequences and conditions for allele-specific PCR are available upon request. We followed a hierarchical typing strategy (Underhill et al. 2000; Hammer et al. 2001), wherein additional genotyping of a sample was restricted to markers on the appropriate branch of the haplogroup tree.
For STR analysis, ten STRs (DYS19, DYS388, DYS389I, DYS389II, DYS390, DYS391, DYS392, DYS393, DYS426, and DYS439) were typed in two multiplex PCR reactions. Primer sequences and PCR conditions have been described previously (Kayser et al. 1997; Redd et al. 2002a). PCR products were electrophoresed on a 3130 Genetic Analyzer (Applied Biosystems, Foster City, CA) using a 36-cm array and filter set D. The data were analyzed with Genescan (v. 3.7, Applied Biosystems) and Genotyper (v. 1.1, Applied Biosystems). We define DYS389CD as equivalent to DYS389I, and we define DYS389AB as equivalent to DYS389II minus DYS389I.
Statistical analyses
For our single nucleotide polymorphism (SNP) data, we used ARLEQUIN 2.0 software (Schneider et al. 2000) to calculate ΦST distances (Excoffier et al. 1992), which include frequency as well as molecular distance information. Y-STR diversity was estimated by using the software package MICROSAT 1.5d (Minch 1997). Multidimensional scaling (MDS) (Kruskal 1964) was performed on the ΦST distances using the software package NTSYS (Rohlf 1998). Median-joining networks (Bandelt et al. 1999) were constructed using the NETWORK 4.0c program. For network calculations, STRs were weighted according to their repeat number variances such that higher weights were assigned to the least variable loci. The reduced median output was used as input for the median-joining network. This procedure reduces the chances of obtaining large reticulations within the network (Hurles et al. 2002).
Estimates of haplogroup ages based on microsatellite diversity were obtained by two methods. We performed Bayesian analyses of coalescent simulations of Y-STR diversity by using BATWING (Wilson and Balding 1998), which is dependent on a population model. We explored three models: (1) constant population size; in this case BATWING implements the standard coalescent time; (2) pure exponential growth at rate alpha to a current population size; and (3) a model that incorporates a population growth at time beta with growth rate alpha. Prior distribution for the STR mutation rate was specified as a log normal with a mean of 6.9×10−4 per locus per generation (Zhivotovsky et al. 2004). For the initial effective population size, we used a log normal prior distribution (5,1). We considered a model of exponential growth with prior distributions as a gamma (1,14) for alpha and a gamma (5,1) for beta parameters. The population size and growth rate parameters were based on data presented by Koyama (1992). Each BATWING run consisted of 20,000 Markov steps after a warm-up of 1,000. We also used the YMRCA method, which is based entirely on genetic parameters such as mutation rates and allele length (Stumpf and Goldstein 2001). The YMRCA method calculates the genetic distance between all current chromosomes and an inferred ancestral haplotype average over all loci. We assumed the same mutation rate as above, and a generation time of 25 years. The confidence limits for the age estimates were calculated with consideration of the 95% confidence interval (CI) of the mutation rate estimates.
Results
Geographic distribution of NRY haplogroups
Y chromosome haplogroup frequencies for 6 Japanese and 33 Asian populations are presented in Fig. 2, which also shows the evolutionary relationships among the 41 haplogroups represented in this survey. A total of 19 binary haplogroups is observed in Japanese populations. Four major haplogroups C, D, N, and O) account for 98.9% of Japanese Y chromosomes, with haplogroups O and D comprising the majority of these (~86.4%). Haplogroup O is the most frequent haplogroup in Japan, accounting for 51.7% of Japanese Y chromosomes in this survey. It is found in all Japanese groups except the Ainu, with frequencies ranging from 37.8% in Okinawa to 46.2–62.3% on mainland Japan (Table 1). There are two major sub-clades of this haplogroup, O-P31 and O-M122 (Fig. 2), both of which are present at high frequencies in Japan. O-P31 is present at a frequency of 31.8% and is split into two sub-clades, O-SRY465 and O-M95 (Fig. 2). O-SRY465* and its derived haplogroup O-47z are almost entirely restricted to, and have opposite frequency patterns in, Japan (7.7 and 22.0%, respectively) and Korea (33.3 and 4.0%, respectively) (supplementary material). O-47z is found in all Japanese populations except the Ainu, with higher frequencies on mainland Japan (21.3–28.3%) than in Okinawa (11.1%). O-M95, a marker found at notable frequencies in Southeast Asia and south Asian tribal populations and rarely in northeast Asia, is present at 1.9% in Japan (supplementary material). O-M122, the other major sub-clade of haplogroup O, is represented in Japan by two derived subtypes, O-M134 (10.4%) and O-LINE (3.1%), as well as by chromosomes not marked by any downstream mutations, O-M122* (6.6%) (Fig. 2). These lineages are very common in southeastern Asia, Oceania, and several central Asian populations, with O-LINE1 and O-M134 comprising almost 50% of all sampled chromosomes in China (supplementary material).

Maximum-parsimony tree of 44 Y chromosome haplogroups together with their frequencies in Japan and five Asian regions. Samples sizes for each region: Japan 259; northeast Asia (NEA) 441; Southeast Asia (SEA) 683; central Asia (CAS) 419; south Asia (SAS) 496; Oceania (OCE) 209. Major clades (i.e., C–R) are labeled with upper case letters to the left of each clade. Mutation names are given along the branches. The length of each branch is not proportional to the number of mutations or the age of the mutation. Dotted lines indicate internal nodes not defined by downstream markers (i.e., paragroups). The names of the 41 haplogroups observed in the present study are shown to the right of the branches. Haplogroup frequencies are shown on the far right, and frequencies of selected Japanese clades are shown within black boxes
The D clade, Japan’s second most frequent haplogroup, is divided into three sub-clades: D-P37.1, D-M15, and D-P47 (Fig. 2). The D-P37.1 lineage is a remarkably long branch on the Y haplogroup tree, with six mutations falling on an internal branch, and three downstream mutations defining three sub-lineages (D-M116.1, D-M125, D-P42). Along with D-P37.1*, these sub-lineages account for 34.7% of Japanese Y chromosomes (Table 1). Haplogroup D exhibits a very different spatial pattern than haplogroup O in Japan, with frequencies varying from 75% in the Ainu to 25.7% in Tokushima (Table 1). Outside of Japan D-P37.1 and its sub-lineages are extremely rare, being found in only three Korean males (D-P37.1* and D-M125*) and one male from Micronesia (D-M116.1*). The other two sub-clades of D-M174 are found only on mainland Asia. D-M15 is present in Tibet, Mongolia, and Southeast Asia, and D-M174* is found in Central Asia. The newly identified P47 mutation establishes a fourth Asian D lineage that marks most chromosomes that were previously ancestral D-M174* from Central Asia (Karafet et al. 2001).
The third most frequent major clade in Japan is haplogroup C, which accounts for 8.5% of Japanese Y chromosomes in this survey (Table 1). Three sub-clades in haplogroup C are marked by the mutations M8, M217, and M38 (Fig. 2). C-M217 and the derived C-M86 lineage are common haplogroups in Northeast and Central Asia, but are present only at low frequency in Japan (1.9 and 1.2%, respectively). Interestingly, C-M217 is found in the Ainu and mainland Japanese, but not in Okinawa (Table 1). C-M38 chromosomes are found only in the Pacific and east Indonesia. C-M8 chromosomes are completely limited to the Japanese archipelago, present at 5.4% on the mainland and Okinawa, but not in the Ainu. Haplogroup N is the fourth most common haplogroup in Japan (1.5%) and is found only among mainland Japanese (Table 1). Clades N and O share a common node in the Y chromosome tree that is defined by marker M214. While NO* chromosomes are extremely rare, they are found in Japan at higher frequency than elsewhere in our survey, albeit only at 2.3%.
Non-metric MDS plots
Figure 3 presents an MDS plot based on ΦST genetic distances, with a low stress value of 0.16. All southeastern Asian populations cluster together on the left side of the plot; with only northern Han Chinese, Korean, and Manchu populations showing closer affinities with southeastern groups than with their geographic neighbors. All other northeast Asians, as well as central Asians, south Asians, and Oceanic populations, are on the right side of the plot. All Japanese populations form a cluster on the upper side of Fig. 3, with the Ainu as an outlier and populations from Kyushu, Tokushima, Shikoku, and Aomori occupying a position closer to the Southeast Asian cluster. The presence of Tibetans within the Japanese cluster is likely due to prevalence of D lineages in both populations (supplementary material).

MDS plot of 39 Japanese and Asian populations based on ΦST genetic distances. For population codes and symbols, see Fig. 1
Median-joining networks of Y-STR haplotypes
Median-joining networks based on Y-STR haplotypes were constructed to examine STR variation within Japanese candidate founding haplogroups. A median-joining network of O-47z STR haplotypes exhibits star-like features with a common Japanese haplotype accounting for 31% of the entire cluster (Fig. 4a). Four of the five non-Japanese O-47z haplotypes (from Korea and Southeast Asia) lie in the middle of the network and appear to be undifferentiated from Japanese haplotypes. A network of O-SRY465* chromosomes (Fig. 4b), shows much higher haplotype differentiation in Korea than in Japan. Two distinctive clusters are revealed in the median-joining network for the O-M95* lineage (Fig. 4c). All Indian haplotypes are clustered together, while another cluster contains haplotypes from Southeast Asia, including those from Japan. In the median-joining network of C-M8 and C* chromosomes (Fig. 4d), C-M8 haplotypes are connected to an Indian C* cluster, which is distinct from simpler star-like Pacific and Southeast Asian clusters.

Median-joining microsatellite networks for lineages O-47z (a), O-SRY465* (b), O-M95* (c), and C* (d) (the position of the M8 mutation is denoted by a black line and the open circle represents a single central Asian haplotype). Microsatellite haplotypes are represented by circles with area proportional to the number of individuals with that haplotype. Branch lengths are proportional to the number of one-repeat mutations separating the two haplotypes. Haplotypes are color-coded by geographic region (see key), with haplotype sharing indicated by pie chart divisions
Haplogroup dating
Table 2 reports the time to the most recent common ancestor (TMRCA) for the three Y chromosome lineages that are almost entirely restricted to Japan. There is good agreement between the BATWING method when constant size model is assumed and the YMRCA method for the age of the D-P37.1 lineage (20,180 and 19,350 years, respectively). This is not the case for the C-M8 and O-47z haplogroups where estimates of 20,010 years vs 11,650 years and 12,270 years vs 7,870 years were returned for the BATWING and YMRCA methods, respectively. Nevertheless, both dating methods yielded considerably older coalescence times for the D-P37.1 and C-M8 haplogroups compared with those for O-47z. BATWING was also used to explore two alternative demographic models (Table 2). Continuous growth has the effect of reducing the coalescence time for the D-P37.1, C-M8, and O-47z lineages (6,290, 9,710, and 3,810 years, respectively), while growth from a constant size population returns intermediate ages for C-M8 (14,510 years) and O-47z (8,690 years), and a similar age as that returned in a constant size model for the D-P37.1 lineage (19,410 years).
Discussion
Japanese founder Y chromosomes and the dual origins of mainland Japanese
This survey of Y chromosome SNPs in Asia reveals a set of 41 haplogroups, 19 of which are present in Japan (Fig. 2). Three haplogroups are almost entirely restricted to the Japanese archipelago: haplogroup D-P37.1 and its descendants (D-P37.1*, D-M116.1*, D.M125*, and D-P42), O-47z, and C-M8. These lineages account for 34.7, 22.0, and 5.4% of Japanese Y chromosomes, respectively, and may have originated on the Japanese archipelago. The Japanese population also has high frequencies of other haplogroup O lineages that are shared mainly with Southeast Asian populations, and C lineages that are shared primarily with northern Asians. In this section, we make the case that these Y chromosome lineages descend from different ancestral populations that gave rise to the Jomon and Yayoi cultures.
More than 10 years ago, Hammer and Horai (1995) surveyed a small number of Y-linked polymorphisms in three Japanese populations and hypothesized that both Jomon and Yayoi male lineages (marked by YAP+ and p47z, respectively) made contributions to the contemporary mainland Japanese population in support of the hybridization model. We tested this hypothesis in the context of spatial patterns predicted by the three competing models for the origin of mainland Japanese. Sokal and Thomson (1998) suggested that if the hybridization model is correct, there should be U-shaped (or inverted U-shaped) clines centered on Kyushu and southern Honshu. They interpreted their finding of alleles at several classical loci with these spatial patterns as diffusion associated with the Yayoi culture. Under the transformation or substitution models no U-shaped clines are expected. We plotted the frequencies of haplogroups D, O-P31, and O-M122 in each of our six Japanese samples against the approximate geographic distances of each of these populations from Kyushu Island (Fig. 5). Together, these haplogroups account for 86.9% of Japanese Y chromosomes. There is a U-shaped cline for haplogroup D, and inverted U-shaped patterns for haplogroups in clade O. The pattern of higher frequencies of haplogroup O in southwestern Japan and haplogroup D lineages among the Ainu and Ryukyuans is concordant with the hybridization model. Based on the frequencies of these two clades, we estimate the Jomon contribution to modern Japanese to be 40.3%, with the highest frequency in the Ainu (75%) and Ryukyuans (60%). On the other hand, Yayoi Y chromosomes account for 51.9% of Japanese paternal lineages, with the highest contribution in Kyushu (62.3%) and lower contributions in Okinawa (37.8%) and northern Honshu (46.2%) (Fig. 5). Interestingly, there is no evidence for Yayoi lineages in the Ainu (Table 1). The presence of C-M217 chromosomes in this population may well be the result of male gene flow from north Asian populations on northern Sakhalin Island and the Kamchatka Peninsula to the Ainu on Hokkaido (Tajima et al. 2004). The very low incidence of D chromosomes in Korea and in Micronesia is likely explained by recent admixture. This is not surprising given that Korea was ruled by Japan from 1910 to 1945 and Micronesia was under Japanese control for more than three decades since1914 until the end of World War II. The same may be true for the very few O-47z chromosomes found in Korea and Southeast Asia.

Haplogroup frequencies for six Japanese samples plotted against their approximate geographic distances from Kyushu. Black line/solid circles Haplogroup D, dotted gray line/open squares haplogroup O-P31, solid gray line/triangles haplogroup O-M122
Absolute dating estimates based on our Y-STR data provide further support for an earlier wave(s) of migration of haplogroups D and C into Japan and a more recent Yayoi expansion of haplogroup O, in concordance with the hybridization model. Coalescence times for the D and C-M8 lineages are ~20,000 years ago, assuming a constant population size. However, populations have clearly grown dramatically during the Jomon (ca. 21,000 b.p. to 400 b.c.) and Yayoi (ca. 400 b.c. to 300 a.d.) periods (Koyama 1992), so this model may be unrealistic. Pure exponential growth is also unlikely to provide a good model for human population size throughout the Jomon and Yayoi periods because recent high growth rates would imply a vanishingly small population size just a few thousand years ago. Koyama (1992) estimated the number of Paleolithic inhabitants to be ~3,000 individuals based on archaeological, ecological, and ethnographic data. Taking into account marked population growth throughout the Jomon period from ~10,000 until about 4,500 b.p., and more explosive growth during the Yayoi and Kofun (ca. 300–600 a.d.) periods (Koyama 1992), a model of exponential growth from a constant-size ancestral population should yield the most realistic dates. Under this model, the D lineage has a coalescence time of ~19,400 years, with an expansion that started ~12,600 years ago (Table 2). The coalescent time of haplogroup C-M8 is estimated to be ~14,500 years ago, with evidence for population expansion starting ~10,820 years ago. We assume that both systems are detecting the same expansion process given the large confidence intervals on our estimates. Therefore, it is quite possible that these two lineages represent a major and a minor component in a single polymorphic Paleolithic founding population.
The continental origins of Japanese Y haplogroups
Given that we have identified putative Y chromosome markers of Jomon (or pre-Jomon) and Yayoi migrations, can we trace the origins of these lineages before they entered Japan? We infer that the Japanese have at least two lineages (D-P37.1 and C-M8) that descend from Paleolithic founders. Evidence from SNPs and STRs (Table 2) suggests that both lineages have great genealogical depth. In the case of haplogroup D, a striking number of point mutations have accumulated on this Japanese lineage (Fig. 2). The divergence of the Japanese D lineage implies a very early period of dispersal into the Japanese archipelago followed by a long period of isolation from populations on the mainland. There is only one other lineage that exhibits more mutations along an internal branch on the Y chromosome haplogroup tree (YCC 2002; Jobling and Tyler-Smith 2003). This branch, A2, is found among the Khoisan of Namibia, a population that also may have been isolated for a very long period of time (Hammer et al. 2001; Wilder et al. 2004).
The highest frequency of continental D lineages is found in central Asia (Fig. 2), especially in Tibet (50.4%). Evidence for shared ancestry between Tibetans and Japanese is seen in the MDS plot (Fig. 3). We hypothesize that the area between Tibet and the Altai Mountains in northwestern China is the primary candidate region for the geographic source of Paleolithic Japanese founding Y chromosomes. Although Tibetans and people from the Altai have D lineages that are differentiated from those in Japan, there are still “ancestral” chromosomes that are not marked by any known mutations on the D lineage in Tibet and the Altai (Fig. 2). Historical records suggest that Tibetan populations were derived from ancient tribes of northwestern China that subsequently moved to the south and admixed with southern natives in the last 3,000 years (Ruofu and Yip 1993; Cavalli-Sforza et al. 1994; Wen et al. 2004). The survival of ancient lineages within haplogroup D in Tibetans and Japanese may well reflect long periods of isolation for both groups. Interestingly, a Y-SNP survey of Andaman Islanders found a very high frequency of haplogroup D-M174* chromosomes in this isolated population that likely descends from Paleolithic Asian ancestors (Thangaraj et al. 2003). Recent expansions and population replacements in Asia, perhaps associated with the spread of agriculture, may have led to the near extinction of haplogroup D in other Asian populations. The other postulated Japanese Paleolithic founding haplogroup, C-M8, is associated with Y-STR haplotypes that are related to Indian and central Asian C chromosomes (Fig. 4d). The presence of NO* chromosomes in Japan also may be an indication of a remnant Tibetan ancestry (Deng et al. 2004). A recent mtDNA study revealed direct connections of Japanese haplotypes with Tibet, parallel to those found for the Y chromosome (Tanaka et al. 2004). Haplogroup M12 is the mitochondrial counterpart of Y chromosome D lineage. This rare haplogroup was detected only in mainland Japanese, Koreans, and Tibetans, with the highest frequency and diversity in Tibet (Tanaka et al. 2004).
Haplogroup O-SRY465 fits basic criteria (Karafet et al. 1999) for a Yayoi founding lineage because it is widespread in (and almost entirely restricted to) both Japan and Korea. Its higher Y-STR diversity in Korea (Fig. 4b; average repeat size variance in Korea is 0.308 vs 0.258 in Japan, data not shown) is consistent with the hypothesis that O-SRY465 tracks male lineages that migrated to Japan from Korea. Geographic distribution data and the median-joining network in Fig. 4a support the hypothesis that the 47z mutation arose on an ancestral O-SRY465 chromosome during early phases of the Yayoi migration. As in the case of haplogroups D and C, the coalescent time for O-47z depends on the demographic model assumed. However, in this case, a model of pure exponential growth seems the most suitable for estimating the age of this lineage. According to Koyama (1992) the population increased rapidly as a result of a stable agricultural food supply during the Yayoi period. The population size exceeded 106 by the end of Yayoi period. This growth continued through historic times, and by the end of the Edo period (ca. 1868) the population is estimated to have grown to a size of 3×107. Our estimate for the age of O-47z under a continuous population growth model is 3,810 (1,640–7,960) years (Table 2), consistent with the hypothesis that this lineage expanded in conjunction with Yayoi culture.
Our data also support the hypothesis that other Y haplogroups, such as lineages within haplogroup O-M122 (i.e., O-M134 and O-LINE), as well as the O-M95 lineage within O-P31, entered Japan with the Yayoi expansion (Fig. 5). High frequencies of these lineages in southwestern Japan, Korea, and Southeast Asian populations likely explain the affinity of these populations in the MDS plot (Fig. 3). The entire O haplogroup has been proposed to have a Southeast Asian origin (Su et al. 1999; Kayser et al. 2000; Capelli et al. 2001; Karafet et al. 2001). In fact, nearly all lineages within the O-M175 clade in Fig. 2, except O-SRY465 and O-47z, are present at their highest frequencies (e.g., O-M95, O-P31*, M122*, O-LINE, O-M119) in southeastern Asia/Oceania (Fig. 2), and have been proposed to have southern Chinese origins (Santos et al. 2000; Su et al. 2000; Karafet et al. 2005). Their expansion into surrounding regions likely accompanied the proliferation of Neolithic culture and rice cultivation. We hypothesize that the dispersals of Neolithic farmers from Southeast Asia also brought haplogroup O lineages to Korea and eventually to Japan.
In summary, our data suggest that Paleolithic male lineages entered Japan at least (12,000–20,000 years ago from central Asia, and were isolated for thousands of years once land bridges between Japan and continental Asia disappeared at the end of the last glacial maximum (~12,000 years ago). More recently, Y chromosomes that originated in Southeast Asia expanded to Korea and Japan with the spread of wet rice agriculture. The ages and spatial patterns of haplogroups D and O in Japan are concordant with the hypothesis that Y chromosomes spread via a process of demic diffusion during the Yayoi period (Sokal and Thomson 1998). Each of the populations carrying these differentiated lineages made separate contributions to modern Japanese, both genetically and culturally. In contrast to previous models, we propose that the Yayoi Y chromosomes descend from prehistoric farmers that had their origins in southeastern Asia, perhaps going back to the origin of agriculture in this region. This places the Yayoi in the context of other population expansions stimulated by the acquisition of agriculture, whereby farming societies gained advantages over hunter-gatherer societies (Diamond and Bellwood 2003). In this case, however, the Jomon hunter-gatherers may have held off the onslaught of farmers for thousands of years as a result of their highly successful brand of subsistence. The dramatic Yayoi transition finally may have been triggered in 400 b.c. by a combination of developments, such as rice field irrigation, cold-resistant rice strains, an increasing Korean population, and the invention of iron tools for producing farming implements (Diamond 1998). The data indicate, however, that Jomon genes survive at high frequencies in contemporary Japanese, possibly because their unique and varied culture complemented that of the immigrant farmers.
References
Aikens CM, Higuchi T (1982) Prehistory of Japan. Academic Press, New York, NY
Bandelt HJ, Forster P, Rohl A (1999) Median-joining networks for inferring intraspecific phylogenies. Mol Biol Evol 16:37–48
Bannai M, Tokunaga K, Imanishi T, Harihara S, Fujisawa K, Juji T, Omoto K (1996) HLA class II alleles in Ainu living in Hidaka District, Hokkaido, northern Japan. Am J Phys Anthropol 101:1–9
Capelli C, Wilson JF, Richards M, Stumpf MP, Gratrix F, Oppenheimer S, Underhill P, Pascali VL, Ko TM, Goldstein DB (2001) A predominantly indigenous paternal heritage for the Austronesian-speaking peoples of insular Southeast Asia and Oceania. Am J Hum Genet 68:432–443
Cavalli-Sforza LL, Menozzi P, Piazza A (1994) The history and geography of human genes. Princeton University Press, Princeton, NJ
Chard C (1974) Northeast Asia in prehistory. University of Wisconsin Press, Madison, WI
Cruciani F, Santolamazza P, Shen PD, Macaulay V, Moral P, Olckers A, Modiano D, Holmes S, Destro-Bisol G, Coia V, Wallace DC, Oefner PJ, Torroni A, Cavalli-Sforza LL, Scozzari R, Underhill PA (2002) A back migration from Asia to sub-Saharan Africa is supported by high-resolution analysis of human Y-chromosome haplotypes. Am J Hum Genet 70:1197–1214
De Knijff P (2000) Messages through bottlenecks: on the combined use of slow and fast evolving polymorphic markers on the human Y chromosome. Am J Hum Genet 67:1055–1061
Deng W, Shi B, He X, Zhang Z, Xu J, Li B, Yang J, Ling L, Dai C, Qiang B, Shen Y, Chen R (2004) Evolution and migration history of the Chinese population inferred from Chinese Y-chromosome evidence. J Hum Genet 49:339–348
Diamond J (1998) Japanese roots. Discover 19:86–94
Diamond J, Bellwood P (2003) Farmers and their languages: the first expansions. Science 300:597–603
Excoffier L, Smouse PE, Quattro JM (1992) Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131:479–491
Hammer MF, Horai S (1995) Y chromosomal DNA variation and the peopling of Japan. Am J Hum Genet 56:951–962
Hammer MF, Zegura SL (2002) The human Y chromosome haplogroup tree: nomenclature and phylogeography of its major divisions. Annu Rev Anthropol 31:303–321
Hammer MF, Karafet TM, Redd AJ, Jarjanazi H, Santachiara-Benerecetti S, Soodyall H, Zegura SL (2001) Hierarchical patterns of global human Y-chromosome diversity. Mol Biol Evol 18:1189–1203
Hammer MF, Blackmer F, Garrigan D, Nachman MW, Wilder JA (2003) Human population structure and its effects on sampling Y chromosome sequence variation. Genetics 164:1495–1509
Hanihara K (1991) Dual structure model for the population history of Japanese. Jpn Rev 2:1–33
Horai S, Murayama K, Hayasaka K, Matsubayashi S, Hatori Y, Fucharoen G, Harihara S, Park KS, Omoto K, Pan I-H (1996) mtDNA polymorphism in East Asian populations, with special reference to the peopling of Japan. Am J Hum Genet 59:579–590
Howells WW (1966) The Jomon people of Japan: a study by discriminant analysis of Japanese and Ainu crania. Papers of the Peabody Museum of Archaeology and Ethnology, Harvard University, 57:1–43
Hurles ME, Nicholson J, Bosch E, Renfrew C, Sykes BC, Jobling MA (2002) Y chromosomal evidence for the origins of oceanic-speaking peoples. Genetics 160:289–303
Jobling MA, Tyler-Smith C (2003) The human Y chromosome: an evolutionary marker comes of age. Nat Rev Genet 4:598–612
Karafet TM, Zegura SL, Posukh O, Osipova L, Bergen A, Long J, Goldman D, Klitz W, Harihara S, de Knijff P, Wiebe V, Griffiths RC, Templeton AR, Hammer MF (1999) Ancestral Asian source(s) of New World Y-Chromosome founder haplotypes. Am J Hum Genet 64:817–831
Karafet T, Xu L, Du R, Wang W, Feng S, Wells RS, Redd AJ, Zegura SL, Hammer MF (2001) Paternal population history of East Asia: sources, patterns, and microevolutionary processes. Am J Hum Genet 69:615–628
Karafet TM, Lansing JS, Redd AJ, Reznikova S, Watkins JC, Surata SPK, Arthawiguna WA, Mayer L, Bamshad M, Jorde LB, Hammer MF (2005) A Balinese Y chromosome perspective on the peopling of Indonesia: genetic contributions from pre-Neolithic hunter-gatherers, Austronesian farmers, and Indian traders. Hum Biol 77:93–114
Kayser M, de Knijff P, Dieltjes P, Krawczak M, Nagy M, Zerjal T, Pandya A, Tyler-Smith C, Roewer L (1997) Applications of microsatellite-based Y chromosome haplotyping. Electrophoresis 18:1602–1607
Kayser M, Brauer S, Weiss G, Underhill PA, Roewer L, Schiefenhovel W, Stoneking M (2000) Melanesian origin of Polynesian Y chromosomes. Curr Biol 10:1237–1246
Koyama S (1992) Prehistoric Japanese populations: a subsistence-demographic approach. In: Japanese as a member of the Asian and Pacific populations. Kyoto, Japan
Kruskal JB (1964) Multidimensional scaling by optimizing goodness of fit to a nonmetric hypothesis. Pyschometrika 29:1–27
Matsumoto T, Imamura O, Yamabe Y, Kuromitsu J, Tokutake Y, Shimamoto A, Suzuki N, Satoh M, Kitao S, Ichikawa K, Kataoka H, Sugawara K, Thomas W, Mason B, Tsuchihashi Z, Drayna D, Sugawara M, Sugimoto M, Furuichi Y, Goto M (1997) Mutation and haplotype analyses of the Werner’s syndrome gene based on its genomic structure: genetic epidemiology in the Japanese population. Hum Genet 100:123–130
Minch E (1997) Microsat. Stanford, UK
Mizoguchi Y (1986) Contributions of prehistoric Far East populations to the population of modern Japan: a Q-mode path analysis based on cranial measurements. In: Akazawa T, Aikens CM (eds) Prehistoric hunter gatherers in Japan, vol Bull No. 27. University of Tokyo Press, Tokyo, pp 107–136
Nei M (1995) The origins of human populations: genetic, linguistic, and archeological data. In: Brenner S, Hanihara K (eds) The original past of modern humans as viewed from DNA. World Scientific, Singapore, pp 71–91
Omoto K, Misawa S (1976) The genetic relations of the Ainu. In: Kirk RL, Thorne AG (eds) The origins of the Australians. The Australian Institute of Aboriginal Studies, Canberra, pp 365–376
Omoto K, Saitou N (1997) Genetic origins of the Japanese: a partial support for the dual structure hypothesis. Am J Phys Anthropol 102:437–446
Ono A, Sato H, Tsutsumi T, Kudo Y (2002) Radiocarbon dates and archaeology of the Late Pleistocene in the Japanese islands. Radiocarbon 44:477–494
Ossenberg N (1986) Isolate conservatism and hybridization in the population history of Japan: the evidence of nonmetric cranial traits. In: Akazawa T, Aikens CM (eds) Prehistoric hunter gatherers in Japan, vol Bull No. 27. University of Tokyo, Tokyo, pp 199–215
Redd AJ, Agellon AB, Kearney VA, Contreras VA, Karafet T, Park H, de Knijff P, Butler JM, Hammer MF (2002a) Forensic value of 14 novel STRs on the human Y chromosome. Forensic Sci Int 130:97–111
Redd AJ, Roberts-Thomson J, Karafet T, Bamshad M, Jorde LB, Naidu JM, Walsh B, Hammer MF (2002b) Gene flow from the Indian subcontinent to Australia: evidence from the Y chromosome. Curr Biol 12:673–677
Rohlf FJ (1998) NTSYS-pc: numerical taxonomy and multivariate analysis system. Sekauket, NY
Ruofu D, Yip VF (1993) Ethnic groups in China. Science Press, Beijing
Santos FR, Pandya A, Kayser M, Mitchell RJ, Liu A, Singh L, Destro-Bisol G, Novelletto A, Qamar R, Mehdi SQ, Adhikari R, de Knijff P, Tyler-Smith C (2000) A polymorphic L1 retroposon insertion in the centromere of the human Y chromosome. Hum Mol Genet 9:421–430
Schneider S, Roessli D, Excoffier L (2000) Arlequin: a software for population genetic analysis. release 1.1, Geneva
Sokal RR, Thomson BA (1998) Spatial genetic structure of human populations in Japan. Hum Biol 70:1–22
Stumpf MP, Goldstein DB (2001) Genealogical and evolutionary inference with the human Y chromosome. Science 291:1738–1742
Su B, Xiao J, Underhill P, Deka R, Zhang W, Akey J, Huang W, Shen D, Lu D, Luo J, Chu J, Tan J, Shen P, Davis R, Cavalli-Sforza L, Chakraborty R, Xiong M, Du R, Oefner P, Chen Z, Jin L (1999) Y-Chromosome evidence for a northward migration of modern humans into Eastern Asia during the last Ice Age. Am J Hum Genet 65:1718–1724
Su B, Xiao C, Deka R, Seielstad MT, Kangwanpong D, Xiao J, Lu D, Underhill P, Cavalli-Sforza L, Chakraborty R, Jin L (2000) Y chromosome haplotypes reveal prehistorical migrations to the Himalayas. Hum Genet 107:582–590
Suzuki H (1981) Racial history of the Japanese. In: Schwidetzky I (ed) Rassengeschichte der Menschheit, 8. Lieferung. Oldenbourg, Munich, pp 7–69
Tajima A, Pan IH, Fucharoen G, Fucharoen S, Matsuo M, Tokunaga K, Juji T, Hayami M, Omoto K, Horai S (2002) Three major lineages of Asian Y chromosomes: implications for the peopling of east and southeast Asia. Hum Genet 110:80–88
Tajima A, Hayami M, Tokunaga K, Juji T, Matsuo M, Marzuki S, Omoto K, Horai S (2004) Genetic origins of the Ainu inferred from combined DNA analyses of maternal and paternal lineages. J Hum Genet 49:187–193
Takamiya H (2002) Peopling of Western Japan, focusing on Kyushu, Shikoku, and Ryukyu Archipelago. Radiocarbon 44:495–502
Tanaka M, Cabrera VM, Gonzalez AM, Larruga JM, Takeyasu T, Fuku N, Guo LJ, et al. (2004) Mitochondrial genome variation in eastern Asia and the peopling of Japan. Genome Res 14:1832–1850
Thangaraj K, Singh L, Reddy AG, Rao VR, Sehgal SC, Underhill PA, Pierson M, Frame IG, Hagelberg E (2003) Genetic affinities of the Andaman Islanders, a vanishing human population. Curr Biol 13:86–93
Turner CG (1976) Dental evidence on the origins of the Ainu and Japanese. Science 193:911–913
Turner CG (1990) Major features of sundadonty and sinodonty, including suggestions about East Asian microevolution, population history and late Pleistocene relationships with Australian aboriginals. Am J Phys Anthropol 82:295–317
Underhill PA, Shen P, Lin AA, Jin L, Passarino G, Yang WH, Kauffman E, Bonne-Tamir B, Bertranpetit J, Francalacci P, Ibrahim M, Jenkins T, Kidd JR, Mehdi SQ, Seielstad MT, Wells RS, Piazza A, Davis RW, Feldman MW, Cavalli-Sforza LL, Oefner PJ (2000) Y chromosome sequence variation and the history of human populations. Nat Genet 26:358–361
Wen B, Xie X, Gao S, Li H, Shi H, Song X, Qian T, Xiao C, Jin J, Su B, Lu D, Chakraborty R, Jin L (2004) Analyses of genetic structure of Tibeto-Burman populations reveals sex-biased admixture in southern Tibeto-Burmans. Am J Hum Genet 74:856–865
Wilder JA, Mobasher Z, Hammer MF (2004) Genetic evidence for unequal effective population sizes of human females and males. Mol Biol Evol 21:2047–2057
Wilson IJ, Balding DJ (1998) Genealogical inference from microsatellite data. Genetics 150:499–510
YCC (2002) A nomenclature system for the tree of Y chromosomal binary haplogroups. Genome Res 12:339–348
Zhivotovsky LA, Underhill PA, Cinnioglu C, Kayser M, Morar B, Kivisild T, Scozzari R, Cruciani F, Destro-Bisol G, Spedini G, Chambers GK, Herrera RJ, Yong KK, Gresham D, Tournev I, Feldman MW, Kalaydjieva L (2004) The effective mutation rate at Y chromosome short tandem repeats, with application to human population-divergence time. Am J Hum Genet 74:50–61
Acknowledgements
We thank Ji Park and Alan Redd for technical assistance and data analysis, and Elizabeth Wood for comments on earlier versions of the manuscript. This research was supported by NSF grant OPP-9806759 to M.F.H.
Author information
Authors and Affiliations
Corresponding author
Additional information
Dedicated to the memory of Satoshi Horai
Electronic Supplementary Material
Rights and permissions
About this article
Cite this article
Hammer, M.F., Karafet, T.M., Park, H. et al. Dual origins of the Japanese: common ground for hunter-gatherer and farmer Y chromosomes. J Hum Genet 51, 47–58 (2006). https://doi.org/10.1007/s10038-005-0322-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-005-0322-0
Keywords
This article is cited by
-
An update and frequency distribution of Y chromosome haplogroups in modern Japanese males
Journal of Human Genetics (2024)
-
Y chromosome analysis for common surnames in the Japanese male population
Journal of Human Genetics (2021)
-
Origin and Spread of the ALDH2 Glu504Lys Allele
Phenomics (2021)
-
Origin of ethnic groups, linguistic families, and civilizations in China viewed from the Y chromosome
Molecular Genetics and Genomics (2021)
-
Population genomics of East Asian ethnic groups
Hereditas (2020)
