
Abstract
Tol2 is an active DNA-based transposable element identified in the medaka fish, Oryzias latipes. Originating from a vertebrate and belonging to the hAT (hobo/Activator/Tam3) transposable element family, featuring a wide distribution among organisms, Tol2 would be expected to be active if introduced into mammals. We, therefore, examined if excision, one part of the transposition reaction, can occur in human and mouse culture cells. A Tol2 clone was introduced into cells and, after incubation, recovered. PCR and sequencing analysis provided evidence for precise and near precise excision in these cells. Tol2 can thus be expected to serve as a material for developing a gene transfer vector and other genetic tools applicable to mammals. It was also suggested that an intact Tol2 element could retain autonomy as a transposable element in mammalian cells.
Similar content being viewed by others

Introduction
DNA-based transposable elements appear to have been nearly or completely inactivated in mammalian genomes, as inferred from results of genome sequencing projects of human and other species. In this situation, an effective approach to develop transposable-element-mediated technologies applicable to mammals would be to use elements from other sources. Elements so far reported to transpose in mammals include Tc1 and Tc3 of Caenorhabidtis elegans (Schouten et al. 1998; Fischer et al. 2001), Himar1 of Haematobia irritans (Zhang et al. 1998), Minos of Drosophila hydei (Zagoraiou et al. 2001) and Sleeping Beauty synthesized on the basis of sequence information for fish elements (Ivics et al. 1997). The last two elements have already contributed to various developments, including transgenesis (Izsvak et al. 2000; Yant et al. 2000), gene tagging (Horie et al. 2001; Dupuy et al. 2002) and promoter trapping (Klinakis et al. 2000). All the elements belong to the mariner/Tc1 transposable element family, members of which are distributed in a wide range of organisms. Another family featuring an even wider distribution is known: the hAT family, which includes hobo of Drosophila, Activator of maize and Tam3 of snapdragon (Calvi et al. 1991; Kempken et al. 2001). Tol2 of the medaka fish, Oryzias latipes, is also included (Koga et al. 1996; Koga and Hori 2001), and its application for a gene transfer vector has already been achieved in fish (Koga et al. 2002). Originating from a vertebrate, this element has great potential as a material for technological applications in mammals. With this goal in mind, the first question to be answered is whether Tol2 is transpositionally active in mammals. This can be readily addressed using mammalian cells in culture, and experimental culture systems, once established, would also be expected to contribute much to further studies using individuals.
DNA-based transposable elements transpose in a "cut-and-paste" fashion (Finnegan 1992). "Cut" and "paste" correspond to excision of the element from chromosome and insertion into a new chromosomal location, respectively. These events can also occur with extrachromosomal DNA (Wirtz et al. 1997). Excision is much easier to detect because loss of a specific copy can be readily manifested with PCR. Detecting insertion requires more complex approaches and an appropriate marker gene. As a prerequisite for detecting insertion, and for the ultimate goal of biotechnology application, we examined, in the present study, if Tol2 can excise in human and mouse culture cells. Excision could in fact be demonstrated to occur, the footprint patterns being similar to those observed in fish. The results also suggest that an intact Tol2 element, containing its transposase gene, can produce a transposase that catalyzes at least excision. One more advance is that a helper plasmid DNA was constructed and this can facilitate excision of a defective Tol2 element.
Materials and methods
Construction of indicator plasmids
The organization of the plasmids we prepared in the present study is shown in Fig. 1.

Organization of plasmids. Nucleotide blocks included as components of the plasmids are indicated with abbreviations for DDBJ sequence files and the nucleotide positions. The abbreviations are: [Tol] D84375 (the Tol2 element), [Tyr] AB010101 (the medaka fish Tyrosinase gene), [Tps] AF013079 (cDNA for the Tol2 transposase), [Egc] U55763 (pEGFP-C1 of Clonetech). The boldface nucleotides are translation start and stop codons in the helper plasmids. The underlined nucleotides are the Kozak sequence for translation initiation. CMV pro. and PA are the CMV promoter and the polyadenylation signal, respectively. The arrows are for exons in the indicator plasmids and reading frames in the helper plasmids. The black triangles are the internal inverted repeats in Tol2. Small open triangles show locations and directions of the PCR primers used in detection assays. The primers are all 28-bp long and represent parts of the sequence of pHSG399 (DDBJ M19087). They were used as primer pairs. The names of the pairs and the nucleotide positions of the primers are: Prec 223–250 and 732–705; Pex1 1,062–1,089 and 1,311–1,284; Pex2 1,134–1,161 and 1,277–1,250
Tol2 was first identified as an insertion sequence in the Tyrosinase gene of an albino mutant fish. A 5.0-kb restriction fragment of the Tyrosinase gene region, including the entire 4.7-kb Tol2 element, was subcloned into pHSG399. This plasmid clone, used as an excision indicator, was designated pInd03.
An internal 1.5-kb region, defined by EcoRV recognition sites, was removed from pInd03, and replaced with a 1.9-kb unrelated DNA fragment. This plasmid, designated pInd03-rep, lacks exon 3 and parts of exons 2 and 4, and is thus deficient in coding for the transposase.
Precise excision of the Tol2 portion from pInd03 and pInd03-rep would be expected to produce a plasmid that contains only the Tol2-flanking regions. Such a plasmid, to be used as a positive control material for excision detection PCR, was artificially prepared by inserting the corresponding region of the wild-type Tyrosinase gene into pHSG399. This plasmid was further treated with restriction enzyme BglII, Klenow enzyme, and T4 DNA ligase, in this order. This treatment abolished a single BglII recognition site contained in the cloned fragment by adding four extra nucleotides. This small change in the sequence is a mark of proof against contamination of assay samples with this plasmid, designated as pCon03.
Construction of helper plasmids
A cDNA for the Tol2 transposase cloned earlier (Koga et al. 1999) contains a reading frame starting at several possible methionine codons. An internal 1.9-kb region, from the second methionine codon to the stop codon, was amplified by PCR. The primer for the 5′ end had been designed to introduce the Kozak signal for translation initiation (Kozak 1987). The PCR product was cloned into pCI (Promega), which carries the CMV promoter and a polyadenylation signal. This plasmid clone, working as a helper by supplying the Tol2 transposase in cells, was called pHel03. Another slightly longer plasmid carrying the reading frame from the first methionine codon was also made and tested. No difference in the transposase activity was observed between this plasmid and pHel03 in preliminary experiments (data not shown). We, therefore, used only pHel03 as a helper plasmid throughout this work.
Another plasmid, expected to serve as a negative control in the helper function, was constructed. The entire coding region of pHel03 was inverted, and the resultant plasmid was named pHel03-inv.
Assay procedures
Human HeLa cells and mouse NIH/3T3 cells were cultured in DMEM with 10% FBS and antibiotics. Incubation was conducted in an incubator set at 37 °C and 5.0% CO2.
Aliquots of 8×105 cells were seeded in 60-mm dishes and incubated for 24 h. Mixtures of plasmid DNAs were introduced into the cells using the PolyFect Transfection Reagent (Qiagen), following the manufacturer's instructions. The ratio of the amount of the indicator plasmid to that of the helper plasmid was 1:29, and the total amount of DNA for each dish was 3.0 μg and 2.5 μg for HeLa and NIH/3T3 cells, respectively. After 24 h of incubation, cells were washed twice with PBS and further incubated in fresh medium without DNA and the transfection reagent for 44–48 h. After washing twice with PBS, plasmid DNA was recovered from the cells by a standard method. DNA from one dish was dissolved in 200 μl of 10 mM Tris (pH 8.0).
Using 10 μl of DNA solution, PCR amplification in a total volume of 50 μl was conducted with primer pairs Prec or Pex1 (see Fig. 1). The PCR conditions with primer pair Prec, to confirm the recovery of plasmid from cells, were: 100 s at 94 °C; 22×(20 s at 94 °C, 20 s at 64 °C, 40 s at 72 °C); 120 s at 72 °C. Pex1 is to detect excision, and the PCR conditions were the same except for the cycle number, which was 34 instead of 22. To check for PCR products, 4 μl of the reaction solution was electrophoresed on a 1.4% agarose gel.
Cloning and sequencing analysis
Using a 1/100 volume of the remaining PCR product as a template, the second round of PCR amplification was performed with primer pair Pex2 (see Fig. 1). The PCR conditions were the same as for Prec. The product was cloned into pBluecript II SK+ and sequenced with the T7 primer using the LI-COR 6000 sequencing machine.
Results
Excision assay
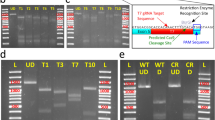
A set of excision assays consisted of four experiments, for all four possible combinations of the two indicator and two helper plasmids. Ten replicated sets were carried out with human cells, and also ten sets with mouse cells. Figure 2 shows agarose gel electrophoresis of the PCR products of the first one set. Primer pair Prec produced fragments of the expected sizes (0.5 kb) in all experiments, indicating that the indicator plasmid DNA was recovered from cells. Product fragments with primer pair Pex1 were seen in the lanes for experiments 1–3, for both human and mouse cells, and their sizes did not differ on the gel from that of pCon03 (0.6 kb). The band intensity was about the same between experiments 1 and 3, and weaker in experiment 2 than in 1 and 3. This pattern was the same through the ten sets, except for experiment 2 with mouse cells. In two of the ten sets with mouse cells, a band was not seen in the lane for experiment 2. We re-examined the PCR products of these two cases after concentration by ethanol precipitation, and found bands of the same size (data not shown). We also treated all the PCR products of experiment 4 in the same way, but could not detect a band (data not shown).

PCR to detect excision. Results of agarose-gel electrophoresis are shown for one set each with human cells and mouse cells. The second lane is a PCR product using 10 pg of pCon03 DNA as template
The appearance of a band in experiment 2 suggests that excision of the Tol2 portion from the indicator plasmid pInd03 occurred, even without pHel03, in human and mouse culture cells. The size of the band was almost the same as that from pCon03, suggesting the excision to be precise or near precise. The stronger bands in experiment 1 than in 2 suggest that the excision frequency is increased by coexistence of pHel03.
Observation of a band in experiment 3 at about the same intensity as in experiment 1 suggests that the transposase encoded by pHel03 is functional both for pInd03 and pInd03-rep. No visible band in experiment 4 indicates that excision did not occur or the excision frequency was lower than in the other experiments, suggesting that pHel03-inv does not supply enough amount of the transposase.
Sequencing analysis
We carried out a second round of PCR amplification with primer pair Pex2, using PCR products of experiment 3 as template, and cloned the products into plasmids. Several clones were available from each dish, but only one clone was chosen at random. This was to ensure that all the clones to be used in the following analyses originated from different cells; i.e., from independent excision events. We sequenced the PCR products to reveal their rearrangement breakpoints. Identical sequences were found, and a total of 20 sequences, ten from human and ten from mouse, were grouped into 11 types. Figure 3 shows their sequences and distribution among the samples.

Nucleotide sequences of PCR products. The top line in the box is the sequence of pInd03. The twenty sequences examined were grouped into 11 types. Their distribution among the human samples and the mouse samples are shown in the two columns on the right. The numbers in parentheses are those of nucleotides present in the sequences but not shown in the figure. TSD stands for target site duplication
In all the samples, all or most of the Tol2 sequence had been removed. The type shown at the top is a precise excision, with loss of the entire Tol2 region and retention of one of the two target site duplication (TSD) units. This type was most frequent in both human and mouse cells. Other types were imprecise, leaving part of the Tol2 terminal regions and/or taking away nucleotides of Tol2-flanking regions. However, whatever the case, excision of Tol2 was clearly demonstrated to occur in human and mouse cells.
Discussion
Tol2 is a DNA-based transposable element naturally occurring in fish genomes, and transposition activity has already been confirmed with fish individuals. In the present study, we could demonstrate that Tol2 can undergo excision in human and mouse cells in culture. Excision is part of the transposition reaction, and hAT family elements have been proposed to transpose in a nonreplicative manner (Kunze 1996). Therefore, it is likely that the entire transposition reaction can proceed in mammalian cells. We are now trying to establish an experimental system to detect insertion.
The excision footprints were found to be similar to those observed in fish (Koga et al. 1996), in that various breakpoints are distributed in and around the TSD sequences. This phenomenon is commonly observed with DNA-based transposable elements (Pohlman et al. 1984). The experimental system we have developed here, using culture cells, can be expected to help clarify factors that determine breakpoints, allowing accurate control of possible influences, such as the expression level of the transposase gene.
A Tol2-specific transposase is required for transposition of Tol2 (Koga and Hori 2000). Taking this into account, the most probable interpretation of the present results would be as follows. Plasmid pHel03 produces the transposase, and pHel03-inv does not. The transposase causes the excision of the Tol2 portion of pInd03 and pInd03-rep. Plasmid pInd03 also produces the transposase, but its production efficiency or the enzyme activity is lower than that of pHel03. While replacement of the internal 1.5-kb region of Tol2 to give pInd03-rep abolishes the production of the transposase, it does not destroy machinery that works as a substrate for the transposase. In most DNA-based elements, such machinery exists in terminal-inverted-repeat sequences and their proximally flanking regions (Fedoroff et al. 1983; O'Hare and Rubin 1983), our results being consistent with this.
Plasmid pInd03 contains an intact Tol2 element, and this was suggested to produce the transposase in the mammalian cells used. Therefore, although a distinct promoter sequence has not been identified in the Tol2 sequence, a promoter that works for the expression of the internal transposase gene is considered to be present. In other words, Tol2 exhibits autonomy with regard to transposition in the mammalian cells used.
In our previous experiments using fish, in vitro synthesized RNA was used to supply the transposase (Koga and Hori 2000; Koga et al. 2002). In the present study, we constructed a helper plasmid, in which the transposase gene is controlled by a strong promoter and some other signals have been added. This plasmid was demonstrated to function at least for excision. This is a methodological improvement because DNA is much easier to handle than RNA.
The present study demonstrated that Tol2 can excise in human and mouse cells. Although excision is part of transposition, its demonstration paves the way for developing a gene tagging system and other genetic tools for mammals using this DNA-based transposable element. This element belongs to the hAT family, and thus has different features from the mariner/Tc1 family elements, some of which have already been applied to the development of genetic tools. One advantage is its larger size. We have already demonstrated that Tol2 can send a DNA fragment that is as large as 9 kb into a fish genome (Koga et al. 2002). Tol2 can thus be expected to serve as a genetic tool as an alternative to mariner/Tc1 elements.
References
Calvi BR, Hong TJ, Findley SD, Gelbert WM (1991) Evidence for a common evolutionary origin of inverted repeat transposons in Drosophila and plants: hobo, Activator, and Tam3. Cell 66:465–471
Dupuy AJ, Clark K, Carlson CM, Fritz S, Davidson AE, Markley KM, Finley K, Fletcher CF, Ekker SC, Hackett PB, Horn S, Largaespada DA (2002) Mammalian germ-line transgenesis by transposition. Proc Natl Acad Sci USA 99:4495–4499
Fedoroff N, Wessler S, Shure M (1983) Isolation of the transposable maize controlling elements Ac and Ds. Cell 35:235–242
Finnegan DJ (1992) Transposable elements. Curr Opin Genet Dev 2:861–867
Fischer SE, Wienholds E, Plasterk RH (2001) Regulated transposition of a fish transposon in the mouse germ line. Proc Natl Acad Sci USA 98:6759–6564
Horie K, Kuroiwa A, Ikawa M, Okabe M, Kondoh G, Matsuda Y, Takeda J (2001) Efficient chromosomal transposition of a Tc1/mariner-like transposon Sleeping Beauty in mice. Proc Natl Acad Sci USA 98:9191–9196
Ivics Z, Hackett PB, Plasterk RH, Izsvák Z (1997) Molecular reconstruction of Sleeping Beauty, a Tc1-like transposon from fish, and its transposition in human cells. Cell 91:501–510
Izsvák Z, Ivics Z, Plasterk RH (2000) Sleeping Beauty, a wide host-range transposon vector for genetic transformation in vertebrates. J Mol Biol 302:93–102
Kempken F, Windhofer F (2001) The hAT family: a versatile transposon group common to plants, fungi, animals, and man. Chromosoma 110:1–9
Klinakis AG, Zagoraiou L, Vassilatis DK, Savakis C (2000) Genome-wide insertional mutagenesis in human cells by the Drosophila mobile element Minos. EMBO Rep 1:416–421
Koga A, Hori H (2000) Detection of de novo insertion of the medaka fish transposable element Tol2. Genetics 156:1243–1247
Koga A, Hori H (2001) The Tol2 transposable element of the medaka fish: an active DNA-based element naturally occurring in a vertebrate genome. Genes Genet Syst 76:1–8
Koga A, Suzuki M, Inagaki H, Bessho Y, Hori H (1996) Transposable element in fish. Nature 383:30
Koga A, Suzuki M, Maruyama Y, Tsutsumi M, Hori H (1999) Amino acid sequence of a putative transposase protein of the medaka fish transposable element Tol2 deduced from mRNA nucleotide sequences. FEBS Lett 461:295–298
Koga A, Hori H, Sakaizumi M (2002) Gene transfer and cloning of flanking chromosomal regions using the medaka fish Tol2 transposable element. Mar Biotechnol 4:6–11
Kozak M (1987) An analysis of 5′-noncoding sequences from 699 vertebrate messenger RNAs. Nucleic Acids Res 15:8125–8148
Kunze R (1996) The maize transposable element Activator (Ac). In: Saedler H, Gierl A (eds) Transposable elements. Springer, Berlin, pp 161–194
O'Hare K, Rubin GM (1983) Structures of P transposable elements and their sites of insertion and excision in the Drosophila melanogaster genome
Pohlman RF, Fedoroff NV, Messing J (1984) The nucleotide sequence of the maize controlling element Activator. Cell 37:635–643
Schouten GJ, van Luenen HG, Verra NC, Valerio D, Plasterk RH (1998) Transposon Tc1 of the nematode Caenorhabditis elegans jumps in human cells. Nucleic Acids Res 26:3013–3017
Wirtz U, Osborne B, Baker B (1997) Ds excision from extrachromosomal geminivirus vector DNA is coupled to vector DNA replication in maize. Plant J 11:125–135
Yant SR, Meuse L, Chiu W, Ivics Z, Izsvák Z, Kay MA (2000) Somatic integration and long-term transgene expression in normal and haemophilic mice using a DNA transposon system. Nat Genet 25:35–41
Zagoraiou L, Drabek D, Alexaki S et al. (2001) In vivo transposition of Minos, a Drosophila mobile element, in mammalian tissues. Proc Natl Acad Sci USA 98:11474–11478
Zhang L, Sankar U, Lampe DJ, Robertson HM Graham FL (1998) The Himar1 mariner transposase cloned in a recombinant adenovirus vector is functional in mammalian cells. Nucleic Acids Res 26:3687–3693
Acknowledgements
We are grateful to D.L. Hartl, S. Hamada and M. Moore for helpful discussion. The HeLa cells were obtained from the Health Science Research Resources Bank of the Japan Health Sciences Foundation. This work was supported by grant no. 10216205 to A.K. and no. 10216206 to A.T. from the Ministry of Education, Culture, Sports, Science and Technology of Japan, and the Basic Science Research Grant from the Sumitomo Foundation to A.K.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Koga, A., Iida, A., Kamiya, M. et al. The medaka fish Tol2 transposable element can undergo excision in human and mouse cells. J Hum Genet 48, 231–235 (2003). https://doi.org/10.1007/s10038-003-0016-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10038-003-0016-4
Keywords
This article is cited by
-
Complete fusion of a transposon and herpesvirus created the Teratorn mobile element in medaka fish
Nature Communications (2017)
-
Enzymatic engineering of the porcine genome with transposons and recombinases
BMC Biotechnology (2007)
-
The Tol1 element of medaka fish is transposed with only terminal regions and can deliver large DNA fragments into the chromosomes
Journal of Human Genetics (2007)
-
Insertional mutagenesis in mice: new perspectives and tools
Nature Reviews Genetics (2005)
